Chemical Reaction
and Molecular Dynamics in Supercritical Fluids
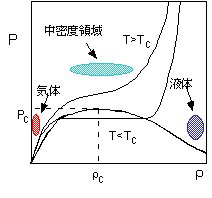 Science using supercritical fluids (SCFs),
which connects gaseous phase and liquid phase continuously, have made a great advance
since the latter half of 1980’s, in various flied including the separation and
extraction, organic synthesis, surface cleaning, creation of microclusters, and so on. The most characteristic feature of SCFs
is the large density fluctuation near the critical point. The central problem in physical
chemistry of SCFs has been how this density fluctuation is reflected in the
various kinds of processes of solute molecules dissolved in SCFs. In general, there is a tendency that the
solvent molecules are close to the solute molecule more densely than the bulk
solvent density, which is called local density enhancement. The effects of the density fluctuation
do not simply appear various kinds of observables, besides some exceptions such
as the partial molal volume of the solute
molecule. The appearance of the
density fluctuation or the local density enhancement is strongly dependent on
the typical scale lengths of the observables.
Science using supercritical fluids (SCFs),
which connects gaseous phase and liquid phase continuously, have made a great advance
since the latter half of 1980’s, in various flied including the separation and
extraction, organic synthesis, surface cleaning, creation of microclusters, and so on. The most characteristic feature of SCFs
is the large density fluctuation near the critical point. The central problem in physical
chemistry of SCFs has been how this density fluctuation is reflected in the
various kinds of processes of solute molecules dissolved in SCFs. In general, there is a tendency that the
solvent molecules are close to the solute molecule more densely than the bulk
solvent density, which is called local density enhancement. The effects of the density fluctuation
do not simply appear various kinds of observables, besides some exceptions such
as the partial molal volume of the solute
molecule. The appearance of the
density fluctuation or the local density enhancement is strongly dependent on
the typical scale lengths of the observables.
Our group has been making
research on the solvent effect in the medium-density region of the SCFs,
especially following four topics.
1. Fluctuation of the solvation and the electron
transfer process.
2. Vibrational energy relaxation process
3. Photo-dissociation and recombination
process
4. Molecular diffusion process.
As experimental methods,
we are using the time-resolved spectroscopy (from sub-ps
to sec) together with other technique such as the resonance Raman
spectroscopy. As the time-resolved
spectroscopy, we mainly use the transient absorption, time-resolved
fluorescence, and transient grating methods.
1.
Fluctuation of Solvation and the Electron Transfer Process

 Although there have been many studies on the electronic transition in
relation with the solvation in SCFs, most of them simply discuss the density
dependence of the absorption or fluorescence peak qualitatively, and/or discuss
the local density enhancement based on the adsorption model. There has been no study on the density
dependence of the intramolecular vibrational
structure, solvent reorganization energy, and the electron transfer rate. By using the resonance Raman
spectroscopy and the pico-femto second transient
absorption spectroscopy, we have made several answers for these questions.
Although there have been many studies on the electronic transition in
relation with the solvation in SCFs, most of them simply discuss the density
dependence of the absorption or fluorescence peak qualitatively, and/or discuss
the local density enhancement based on the adsorption model. There has been no study on the density
dependence of the intramolecular vibrational
structure, solvent reorganization energy, and the electron transfer rate. By using the resonance Raman
spectroscopy and the pico-femto second transient
absorption spectroscopy, we have made several answers for these questions.
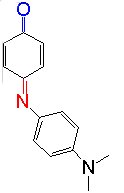
 For
examples, we measured the absorption and resonance Raman spectra of Phenol Blue
(PB), which is a typical solvatochromic dye, in
various solvent fluids including SCFs.
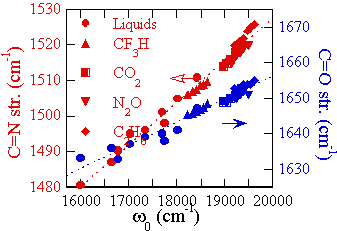
As a result, we have found for the first time the linear relation ship
between the solvent shifts and fluctuations of the electronic transition
energies and vibrational frequencies ( The figure shows the absorption peak and the Ranam Stokes-shift.)
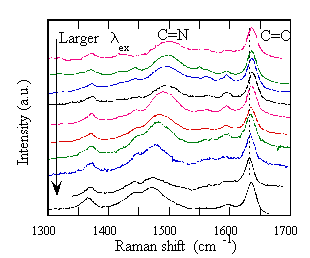
Further we have found a novel phenomena that the Raman Stokes-shift
changes with the change of the excitation wavelength ( The
figure shows the excitation energy ( from 457.9 nm to 647 nm) dependence of the
Raman spectra in methanol. The
study using the SCFs, this phenomenon is revealed to be related to the relation
between the fluctuations of the electronic transition energy and the vibrational frequency.
We also succeeded to explain the experimental results based on this
model. Using these
information, we have discussed the lifetime of the electronic excited state.
For
examples, we measured the absorption and resonance Raman spectra of Phenol Blue
(PB), which is a typical solvatochromic dye, in
various solvent fluids including SCFs.
As a result, we have found for the first time the linear relation ship
between the solvent shifts and fluctuations of the electronic transition
energies and vibrational frequencies ( The figure shows the absorption peak and the Ranam Stokes-shift.)
Further we have found a novel phenomena that the Raman Stokes-shift
changes with the change of the excitation wavelength ( The
figure shows the excitation energy ( from 457.9 nm to 647 nm) dependence of the
Raman spectra in methanol. The
study using the SCFs, this phenomenon is revealed to be related to the relation
between the fluctuations of the electronic transition energy and the vibrational frequency.
We also succeeded to explain the experimental results based on this
model. Using these
information, we have discussed the lifetime of the electronic excited state.
 We have
also determined the back-electron transfer rate of the charge transfer (CT)
complex of hexamethylbenzene and tetracyanoethylene
in various SCFs for the first time by using the ps
transient absorption method.
According to the study of the resonance Raman spectroscopy in liquid
carbon tetrachloride, this CT complex shows peculiar behavior in non-polar
solvent ( large solvent reorganization energy even in
non-polar solvent). By surveying
the density dependence of the absorption spectrum and the back electron
transfer rate over the wide density region from the gaseous region to the
liquid-like region,
we can make a detailed study on the effect of the solvent
reorganization energy in fluids.
According to the experimental results, the reaction rate is well
explained by the solvent reorganization energy and the reaction free energy
estimated from the analysis of the absorption spectrum based on the Marcus and Jortner theory.
In non-polar SCFs, the solvent reorganization energy is not so large and
there is no distinct effect of the density fluctuation.
We have
also determined the back-electron transfer rate of the charge transfer (CT)
complex of hexamethylbenzene and tetracyanoethylene
in various SCFs for the first time by using the ps
transient absorption method.
According to the study of the resonance Raman spectroscopy in liquid
carbon tetrachloride, this CT complex shows peculiar behavior in non-polar
solvent ( large solvent reorganization energy even in
non-polar solvent). By surveying
the density dependence of the absorption spectrum and the back electron
transfer rate over the wide density region from the gaseous region to the
liquid-like region,
we can make a detailed study on the effect of the solvent
reorganization energy in fluids.
According to the experimental results, the reaction rate is well
explained by the solvent reorganization energy and the reaction free energy
estimated from the analysis of the absorption spectrum based on the Marcus and Jortner theory.
In non-polar SCFs, the solvent reorganization energy is not so large and
there is no distinct effect of the density fluctuation.
 On the
other hand, in order to explain the density dependence of the solvent
reorganization energy experimentally observed, we have made calculations of the
solvent reorganization energy using the integral equation theory based on the
interaction site model. Actually, we calculated the difference of the solvation
free energy (DA) and the vertical transition energy (DUt), when the dipole moment of the solute molecule
(dumbbell molecule) changes from 1.4 D to 4.2 D in the solvent fluid of LJ
dumbbell with the dipole of 1.4 D.
As a result, we have succeeded in qualitatively reproducing the density
dependence of the absorption peak shift.
In order to study the solvation structure at each solvent density, we
have calculated the strength of the reaction field acting on the solute
molecule at various dipole moments.
The results are shown in the figure, where the strength of the reaction
field is scaled by the solvent density.
Above the reduced density of 0.5, the reaction field is linear function
of the solute dipole moment. On the
other hand, in the lower density region, the reaction field shows a strong
non-linearity. Further the strength
of the reaction field per solvent molecule is also dependent on the solvent
density. This suggests that the
solvation energy due to
the solvent molecule at the local density around solute is
different at different densities.
This is mainly due to the packing effect at the higher density, which
restricts the solvent orientation.
These results strongly deny the simple adsorption model in interpreting
the density dependence of the absorption spectrum. To ensure these effects, we have also
made detailed study on the fluorescence Stokes-shift of Coumarin
molecule in SCFs.
On the
other hand, in order to explain the density dependence of the solvent
reorganization energy experimentally observed, we have made calculations of the
solvent reorganization energy using the integral equation theory based on the
interaction site model. Actually, we calculated the difference of the solvation
free energy (DA) and the vertical transition energy (DUt), when the dipole moment of the solute molecule
(dumbbell molecule) changes from 1.4 D to 4.2 D in the solvent fluid of LJ
dumbbell with the dipole of 1.4 D.
As a result, we have succeeded in qualitatively reproducing the density
dependence of the absorption peak shift.
In order to study the solvation structure at each solvent density, we
have calculated the strength of the reaction field acting on the solute
molecule at various dipole moments.
The results are shown in the figure, where the strength of the reaction
field is scaled by the solvent density.
Above the reduced density of 0.5, the reaction field is linear function
of the solute dipole moment. On the
other hand, in the lower density region, the reaction field shows a strong
non-linearity. Further the strength
of the reaction field per solvent molecule is also dependent on the solvent
density. This suggests that the
solvation energy due to
the solvent molecule at the local density around solute is
different at different densities.
This is mainly due to the packing effect at the higher density, which
restricts the solvent orientation.
These results strongly deny the simple adsorption model in interpreting
the density dependence of the absorption spectrum. To ensure these effects, we have also
made detailed study on the fluorescence Stokes-shift of Coumarin
molecule in SCFs.
2. Vibrational Energy Relaxation Process
The
understanding of the mechanism of the vibrational
energy relaxation (VER) in solution is on of the central problems in solution
physical chemistry, and there have been so many experiments and theories. Isolated binary collision (IBC) model is
the oldest ones, which is based on the VER in gaseous phase. In this model, VER rate is expressed by
the product of collision frequency (Z) and the energy loss per collision (DE). Although there have been many studies to
elucidate the validity of the IBC model, it is still in question, mainly due to
the difficulty in determining the collision frequency in solution. To overcome this difficulty, we have
studied the VER processes at different electronic states of the same molecule.
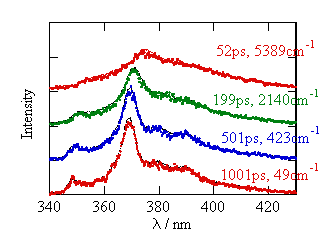 Azulene
is a molecule which emits the fluorescence from the S2 state and the
lineshape of the fluorescence is dependent on the
excess energy. Therefore, it is
possible to examine the VER rate from the lineshape
analysis of the time-resolved fluorescence. The figure shows the fluorescence
spectra at various delay times after the photo-excitation at 283 nm in
ethane. As is shown in the figure,
the fluorescence lineshape become structured with
time, and this change corresponds to the decay of the excess energy.
Azulene
is a molecule which emits the fluorescence from the S2 state and the
lineshape of the fluorescence is dependent on the
excess energy. Therefore, it is
possible to examine the VER rate from the lineshape
analysis of the time-resolved fluorescence. The figure shows the fluorescence
spectra at various delay times after the photo-excitation at 283 nm in
ethane. As is shown in the figure,
the fluorescence lineshape become structured with
time, and this change corresponds to the decay of the excess energy.
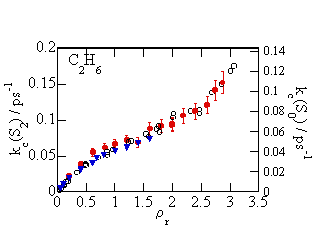 By
comparing the VER rate it the S2 state in ethane thus estimated with
the VER rate in the S0 state in literature, it has been revealed
that the density dependence of the VER rate is quite similar to each
other. However, comparing at the
same solvent density, the VER rate in the S2 state is about 1.5
times faster than that in the S0 state. Similar results are obtained both in
carbon dioxide and xenon. These
results do not contradict to the IBC model. That is, the density dependence of the
VER rate is determined by the collision frequency, and the difference of the
VER rate between ground and excited states are determined by the difference of
the energy loss per collision at different electronic states.
By
comparing the VER rate it the S2 state in ethane thus estimated with
the VER rate in the S0 state in literature, it has been revealed
that the density dependence of the VER rate is quite similar to each
other. However, comparing at the
same solvent density, the VER rate in the S2 state is about 1.5
times faster than that in the S0 state. Similar results are obtained both in
carbon dioxide and xenon. These
results do not contradict to the IBC model. That is, the density dependence of the
VER rate is determined by the collision frequency, and the difference of the
VER rate between ground and excited states are determined by the difference of
the energy loss per collision at different electronic states.
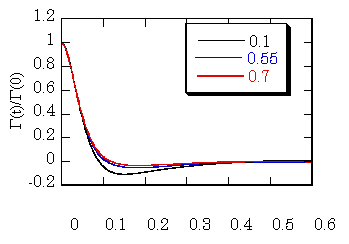 In the
system of weak coupling with solvent, the VER rate is determined by frequency
component (w) of the
real part of the Fourier transform of the time correlation function (G(t))
of the fluctuation force (F(t))acting on the vibrational
coordinate. Since, in general, the vibrational frequency is larger than the solvent motions,
the initial part if the frictional correlation function G(t) is
effective to the vibrational energy relaxation. Therefore, if the functional form of G(t)
is not strongly dependent on the solvent density, the IBC model is applicable
to interpret the density dependence of the VER rate.
In the
system of weak coupling with solvent, the VER rate is determined by frequency
component (w) of the
real part of the Fourier transform of the time correlation function (G(t))
of the fluctuation force (F(t))acting on the vibrational
coordinate. Since, in general, the vibrational frequency is larger than the solvent motions,
the initial part if the frictional correlation function G(t) is
effective to the vibrational energy relaxation. Therefore, if the functional form of G(t)
is not strongly dependent on the solvent density, the IBC model is applicable
to interpret the density dependence of the VER rate.
To clarify this point, we have made a simple calculation. Actually, we have calculated the
fluctuation force acting on the solute molecule interacting with the solvent
molecule with the Lennard-Jones (LJ) potential in the LJ solvent. The temperature of the solvent is 1.5 in
the reduced unit, and the solute-solvent attractive interaction is twice of
that between solvent molecules. The
figure shows the results at different three solvent densities. As is shown in the figure, the
functional form of the initial part is not dependent on the solvent
density. This suggest that the
possibility of the IBC model. We
have also made calculation of the fluctuation due to the repulsive force, and
found that there is no correlation between solvent molecules in the region of
the repulsive force. This makes the
background of the validity of the IBC model.
Now we are
making research from the solvent position using the transient grating
technique.
3.
Photo-Dissociation and Recombination Process
 Let us
consider the case where a molecules composed of two
atoms undergoes photo-dissociation in fluids. The quantum yield of the
photo-dissociation of this molecules is discussed in terms of the potential
energy surface f this molecule and the cage effect due to the surrounding
solvent molecules. Generally the
quantum yield will be expected to decrease with increasing the packing around
the molecule. Now the question is
how the local density enhancement around the solute molecule in the
medium-density region of SCFs affect the quantum
yield. This problem has already
been studied in earlier days, and the German group made extensive studies on
the photo-dissociation quantum yield of iodine in earlier 1980’s. As a result, there has been observed a
extreme decrease of the photo-dissociation quantum yield in carbon dioxide and ethane(J. -C. Dutoit et al., J.
Chem. Phys. 1983, 78, 1825; Luther et al., J. Phys.
Chem. 1980, 84, 3072).
To make sure this observation we have measured the photo-dissociation
quantum yield of iodine by using the transient grating technique.
Let us
consider the case where a molecules composed of two
atoms undergoes photo-dissociation in fluids. The quantum yield of the
photo-dissociation of this molecules is discussed in terms of the potential
energy surface f this molecule and the cage effect due to the surrounding
solvent molecules. Generally the
quantum yield will be expected to decrease with increasing the packing around
the molecule. Now the question is
how the local density enhancement around the solute molecule in the
medium-density region of SCFs affect the quantum
yield. This problem has already
been studied in earlier days, and the German group made extensive studies on
the photo-dissociation quantum yield of iodine in earlier 1980’s. As a result, there has been observed a
extreme decrease of the photo-dissociation quantum yield in carbon dioxide and ethane(J. -C. Dutoit et al., J.
Chem. Phys. 1983, 78, 1825; Luther et al., J. Phys.
Chem. 1980, 84, 3072).
To make sure this observation we have measured the photo-dissociation
quantum yield of iodine by using the transient grating technique.
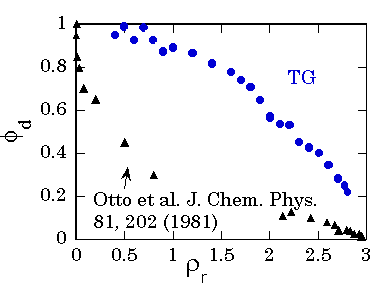 Since the
grating technique can detect the heat during the reaction, it is possible to
determine the photo-dissociation quantum yield by comparing the fast heat due
to the geminate recombination and the slow heat due to the non-geminate
recombination. The results of
the transient grating experiment did not reproduce the anomalous decrease of
the quantum yield in the lower density region. This difference is still an open
problem.
Since the
grating technique can detect the heat during the reaction, it is possible to
determine the photo-dissociation quantum yield by comparing the fast heat due
to the geminate recombination and the slow heat due to the non-geminate
recombination. The results of
the transient grating experiment did not reproduce the anomalous decrease of
the quantum yield in the lower density region. This difference is still an open
problem.
Besides iodine, we are
making research of disulfide compound in SCFs.
4.
Molecular Diffusion Process
See
the page by Ohmori.