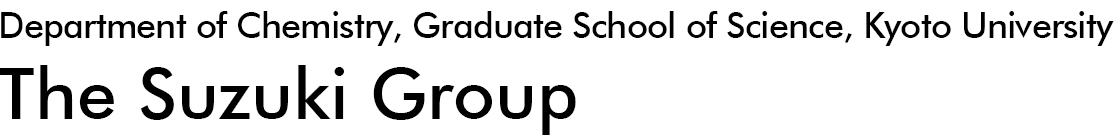
- 1. Ultrafast Photoelectron Spectroscopy of Liquids
- 2. Ultrafast Photoelectron Spectroscopy of Isolated Molecules
- 3. Transient Absorption Spectroscopy
- 4. Development of Ultrafast Lasers
1. Ultrafast Photoelectron Spectroscopy of Liquids
Decoherence and Relaxation of Vibrational Motion in Ultrafast Electronic Relaxation of Pyrazine in Solution
Shutaro Karashima, Tatsuya Ishiyama, and Toshinori Suzuki
J. Phys. Chem. Lett., 17, 410-416 (2026)
We employed extreme-ultraviolet time-resolved photoelectron spectroscopy to investigate the decoherence and relaxation of vibrational motion of pyrazine in aqueous and ethanol solutions. Pyrazine has well-characterized vibronic interactions between the 1B2u (ππ*) and 1B3u (nπ*) states, while recent theoretical and experimental studies have revealed that the 1Au (nπ*) state also participates in the dynamics, inspiring strong interest in electronic and vibrational coherence involving both the 1B3u and 1Au states. Our results indicate that vibrational coherence in water and ethanol persists for approximately 300 fs, similar to that in the gas phase, suggesting that periodic electronic population transfer between the 1B3u and 1Au states may also occur in solution.
Charge Separation Dynamics of Aromatic Molecules at Aqueous Interfaces Revealed by Ultrafast Photoelectron Spectroscopy
Yo-ichi Yamamoto and Toshinori Suzuki
J. Am. Chem. Soc., 148, 1524-1537 (2026)
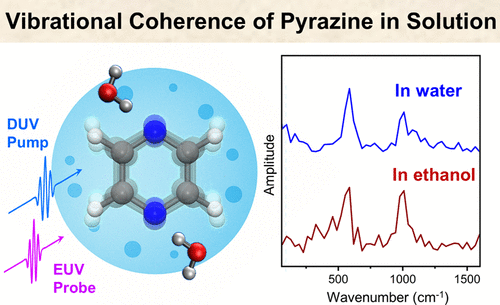
Hydrophobic molecules can preferentially accumulate at the air–water interface, leading to interfacial concentrations far exceeding those in bulk solution and causing reactions to occur at higher rates. However, it remains unclear whether the reaction rate constants are intrinsically altered by the interfacial solvation environment. Here, we employ extreme ultraviolet time-resolved photoelectron spectroscopy (EUV-TRPES) to investigate the photoinduced charge-separation dynamics of the surface-active organic molecules indole, phenol, and phenolate. Ultraviolet photoexcitation of interfacial indole was found to produce both valence excited states and electron–cation contact pairs, both of which decayed faster than in the bulk solution. In contrast, interfacial phenol produced only valence excited states, with no detectable hydrated electrons or their precursors. Interfacial phenolate exhibited faster photodetachment than in the bulk solution. Both indole and phenol exhibit concentration-dependent dynamics due to interfacial molecular aggregation. These results contribute to a more comprehensive understanding of interfacial dynamics when considered alongside prior nonlinear optical spectroscopy studies. We further demonstrate EUV-TRPES with 18 fs time resolution, enabling direct observation of the 5 fs internal conversion of interfacial indole from the 1La to 1Lb state, followed by clear vibrational quantum beats. Thus, EUV-TRPES provides a powerful means for probing electronic and vibrational dynamics at interfaces.
GasLiquid Interface of Aqueous Solutions of Surface Active Aromatic Molecules Studied Using Extreme Ultraviolet Laser Photoelectron Spectroscopy and Molecular Dynamics Simulation
Yo-ichi Yamamoto, Tomonori Hirano, Tatsuya Ishiyama, Akihiro Morita, and Toshinori Suzuki
J. Am. Chem. Soc., 147, 4026-4037 (2025)
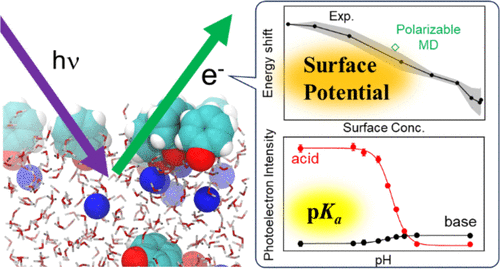
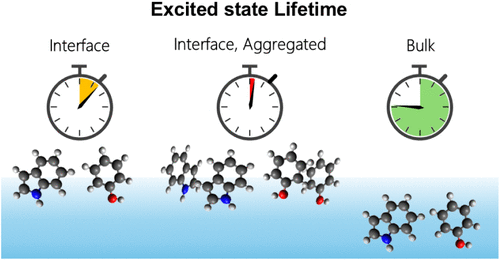
We investigated the gas–liquid interface of aqueous solutions containing phenol and related aromatic compounds using extreme ultraviolet laser photoelectron spectroscopy and molecular dynamics simulations. The interfacial densities of protonated and deprotonated forms of phenol, aniline, and 4-nitrophenol were found to be primarily determined by their surface affinities and exhibit similar concentration dependences to their respective bulk densities. Despite the distinct interfacial orientations of their permanent dipole moments, these compounds monotonically decreased the surface potential at higher concentrations. The exception was sodium phenolate, whose surface potential first increased and then decreased with increasing concentration. This behavior is attributed to the opposing effects of the electric double layer formed by Na+ and phenolate ions at the gas–liquid interface and the induced electronic polarization of phenolate
Exploring the Vibrational Coherences in the Ultrafast Electronic Relaxation of Pyrimidine Nucleobases and Nucleosides
Shutaro Karasshima and Toshinori Suzuki
J. Am. Chem. Soc., 147, 2291-2295 (2025)
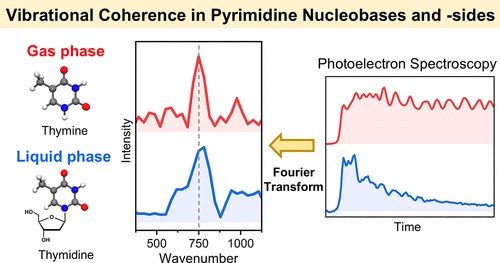
We studied the vibrational coherences during the ultrafast internal conversions (ICs) of pyrimidine nucleobases and -sides in aqueous solutions and the gas phase with an instrumental resolution of 14 fs. The coherence of the same ring-breathing vibrational mode with a frequency of 750 cm-1 was observed. In the gas phase, the vibrational coherence was transferred during IC from the 1ππ* to the 1nπ* state, and it survived for approximately 1 ps. In an aqueous solution, the vibrational dynamics develop in the 1ππ* state because the 1nπ* state is energetically upshifted via hydrogen bonding with H2O, and the vibrational coherence is lost on a similar time scale as that of IC to the ground electronic state
Charge Transfer Reaction at Gas-Liquid Interface of Aqueous Tetrabutylammonium Iodide Solution: Influence of Ions on Dynamical Response of Solvent
BCSJ Award
Shutaro Karashima, Yoshi-Ichi Suzuki, Yo-ichi Yamamoto, and Toshinori Suzuki
Bull. Chem. Soc. Jpn., 97,uoad012 (2024)
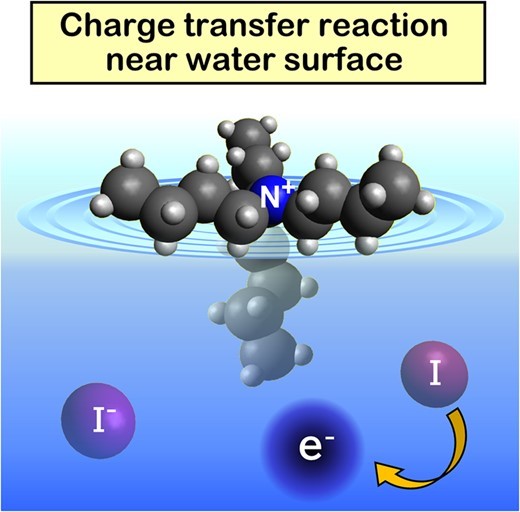
Tetrabutylammonium iodide (TBAI) is known to create an electric double layer at the air-water interface. In the present study, we investigated the charge-transfer-to-solvent reaction from iodide ions to liquid water near the gas-liquid interface of an aqueous TBAI solution using extreme UV (EUV) time-resolved photoelectron spectroscopy. Use of EUV radiation ensured accurate measurements of electron kinetic energy distributions by minimizing spectral distortions caused by electron inelastic scattering in the liquid and the influence of the electron transmission efficiency through the gas-liquid interface. The spectra observed for photodetached electrons exhibited a rapid energy shift and a variation of the bandwidth in subpicoseconds, and the rates of these changes clearly depended on the TBAI concentration. The results indicate that the dynamical response of solvent water changes in the presence of a high density of ions.
Ultrafast Geminate Recombination Facilitated by Hydrogen-Atom Transfer in Charge Transfer Reactions from Hydroxide and Methoxide Ions
Yo-ichi Yamamoto and Toshinori Suzuki
J. Phys. Chem. Lett., 14, 10463-10468 (2023)
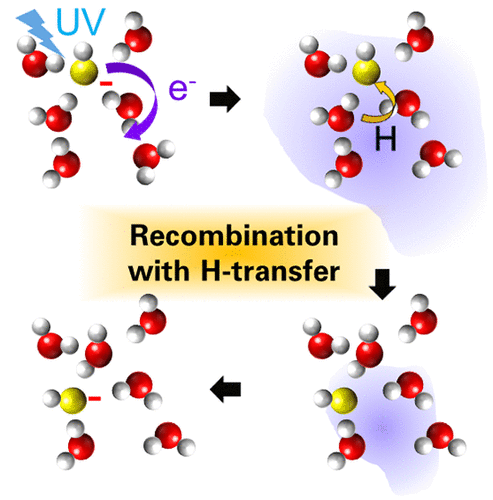
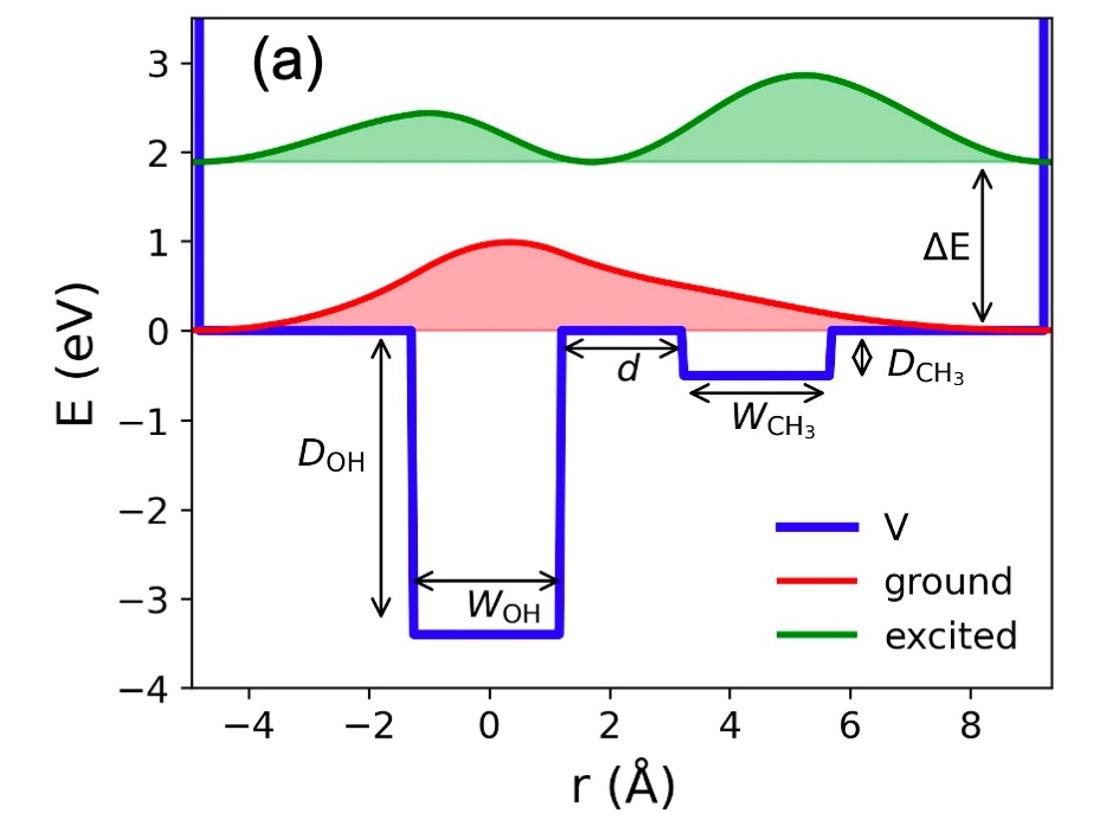
Previous transient absorption spectroscopy (TAS) hinted at an exceptionally rapid geminate recombination process in charge transfer reactions involving OH- or OD- ions in liquid water and CH3O- ions in liquid methanol. However, a comprehensive investigation of these dynamics using TAS has been hindered by the technical challenges stemming from the ultrafast spectral shift that spans a wide wavelength range from the mid-infrared to the visible on the subpicosecond time scale. To address these challenges, we have employed ultraviolet time-resolved photoelectron spectroscopy of aqueous solutions, enabling us to observe and analyze the complete dynamics, including electron detachment, solvation, and geminate recombination. Our findings are consistent with those of Iglev et al. (J. Phys. Chem. Lett. 2015, 6, 986−992), supporting the hypothesis that the structural diffusion of OH/OD/CH3O induced by a presolvated electron plays a pivotal role in facilitating ultrafast geminate recombination.
Extreme Ultraviolet Laser Photoelectron Spectroscopy of Flat Liquid Jet Generated Using Microfluidic Device
BCSJ Award
Yo-ichi Yamamoto, Hiroto Yano, Shutaro Karashima, Ryuta Uenishi, Natsumi Orimo, Junichi Nishitani, and Toshinori Suzuki
Bull. Chem. Soc. Jpn., 96, 938–942(2023)
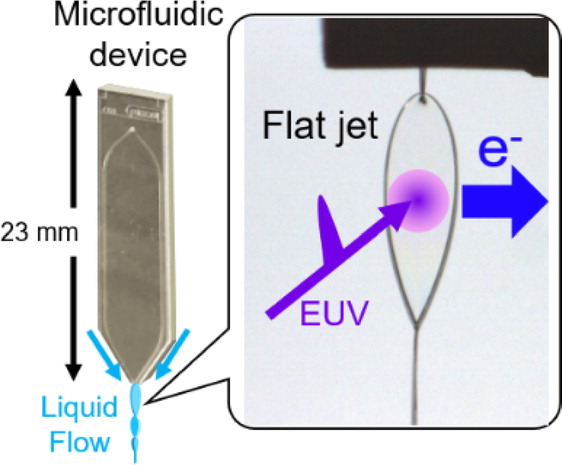
We present photoelectron spectroscopy of liquid films generated in a vacuum with microfluidic devices based on liquid-liquid or gas-liquid collisions. The results are compared to those for a standard liquid microjet technique.
Distortion Correction of Low-Energy Photoelectron Spectra of Liquids Using Spectroscopic Data for Solvated Electrons
Yo-ichi Yamamoto and Toshinori Suzuki
J. Phys. Chem. A., 127, 2440–2452 (2023)
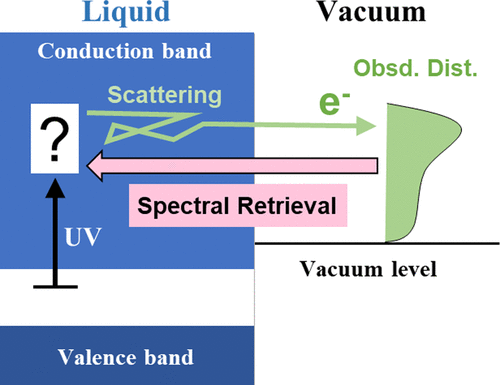
Time-resolved photoelectron spectroscopy (TRPES) enables real-time observation of ultrafast electronic dynamics in solutions. When extreme ultraviolet (EUV) probe pulses are employed, they can ionize solutes from all electronic states involved in the dynamics. However, EUV pulses also produce a strong ionization signal from a solvent that is typically 6 orders of magnitude greater than the pump–probe photoelectron signal of solutes. Alternatively, UV probe pulses enable highly sensitive and selective observation of photoexcited solutes because typical solvents such as water are transparent to UV radiation. An obstacle in such UV-TRPES measurements is spectral distortion caused by electron scattering and a yet to be identified mechanism in liquids. We have previously proposed the spectral retrieval (SR) method as an a posteriori approach to removing the distortion and overcoming this difficulty in UV-TRPES; however, its accuracy has not yet been verified by comparison with EUV-TRPES results. In the present study, we perform EUV-TRPES for charge transfer reactions in water, methanol, and ethanol, and verify SR analysis of UV-TRPES. We also estimate a previously undetermined energy-dependent intensity factor and expand the basis sets for SR analysis. The refined SR method is employed for reanalyzing the UV-TRPES data for the formation and relaxation dynamics of solvated electrons in various systems. The electron binding energy distributions for solvated electrons in liquid water, methanol, and ethanol are confirmed to be Gaussian centered at 3.78, 3.39, and 3.25 eV, respectively, in agreement with Nishitani et al. [Sci. Adv. 2019, 5, eaaw6896]. An effective energy gap between the conduction band and the vacuum level at the gas–liquid interface is estimated to be 0.2 eV for liquid water and 0.1 eV for methanol and ethanol.
Ultrafast Electronic Relaxation in 6-Methyluracil and 5-Fluorouracil in Isolated and Aqueous Conditions: Substituent and Solvent Effects
Natsumi Orimo, Yo-ichi Yamamoto, Shutaro Karashima, Alexie Boyer, and Toshinori Suzuki
J. Phys. Chem. Lett., 14, 2758–2763 (2023)
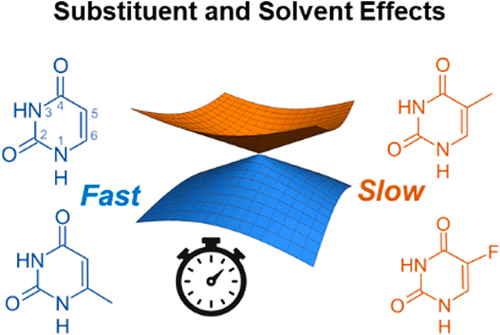
We report ultrafast extreme ultraviolet photoelectron spectroscopy of 6-methyluracil (6mUra) and 5-fluorouracil (5FUra) in the gas phase and 6mUra and 5-fluorouridine in an aqueous environment. In the gas phase, internal conversion (IC) occurs from 1ππ* to 1nπ* states in tens of femtoseconds, followed by intersystem crossing to the 3ππ* state in several picoseconds. In an aqueous solution, 6mUra undergoes IC almost exclusively to the ground state (S0) in about 100 fs, which is essentially the same process as that for unsubstituted uracil, but much faster than that for thymine (5-methyluracil). The different dynamics for C5 and C6 methylation suggest that IC from 1ππ* to S0 is facilitated by out-of-plane (OOP) motion of the C5 substituent. The slow IC for C5-substituted molecules in an aqueous environment is ascribed to the solvent reorganization that is required for this OOP motion to occur. The slow rate for 5FUrd may arise in part from an increased barrier height due to C5 fluorination.
Formation of Long-Lived Dark States during Electronic Relaxation of Pyrimidine Nucleobases Studied Using Extreme Ultraviolet Time-Resolved Photoelectron Spectroscopy
Yuta Miura, Yo-ichi Yamamoto, Shutaro Karashima, Natsumi Orimo, Ayano Hara, Kanae Fukuoka, Tatsuya Ishiyama, and Toshinori Suzuki
J. Am. Chem. Soc., 145, 3369-3381 (2023)
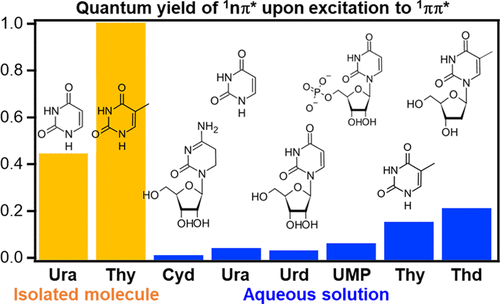
Ultrafast electronic relaxation of nucleobases from 1ππ* states to the ground state (S0) is considered essential for the photostability of DNA. However, transient absorption spectroscopy (TAS) has indicated that some nucleobases in aqueous solutions create long-lived 1nπ*/3ππ* dark states from the 1ππ* states with a high quantum yield of 0.4–0.5. We investigated electronic relaxation in pyrimidine nucleobases in both aqueous solutions and the gas phase using extreme ultraviolet (EUV) time-resolved photoelectron spectroscopy. Femtosecond EUV probe pulses cause ionization from all electronic states involved in the relaxation process, providing a clear overview of the electronic dynamics. The 1nπ* quantum yields for aqueous cytidine and uracil (Ura) derivatives were found to be considerably lower (<0.07) than previous estimates reported by TAS. On the other hand, aqueous thymine (Thy) and thymidine exhibited a longer 1ππ* lifetime and a higher quantum yield (0.12–0.22) for the 1nπ* state. A similar trend was found for isolated Thy and Ura in the gas phase: the 1ππ* lifetimes are 39 and 17 fs and the quantum yield for 1nπ* are 1.0 and 0.45 for Thy and Ura, respectively. The result indicates that single methylation to the C5 position hinders the out-of-plane deformation that drives the system to the conical intersection region between 1ππ* and S0, providing a large impact on the photophysics/photochemistry of a pyrimidine nucleobase. The significant reduction of 1nπ* yield in aqueous solution is ascribed to the destabilization of the 1nπ* state induced by hydrogen bonding.
Charge Transfer Reactions from I– to Polar Protic Solvents Studied Using Ultrafast Extreme Ultraviolet Photoelectron Spectroscopy
Yo-ichi Yamamoto, Yoshi-Ichi Suzuki, and Toshinori Suzuki
J. Phys. Chem. Lett., 14, 1052–1058 (2023)
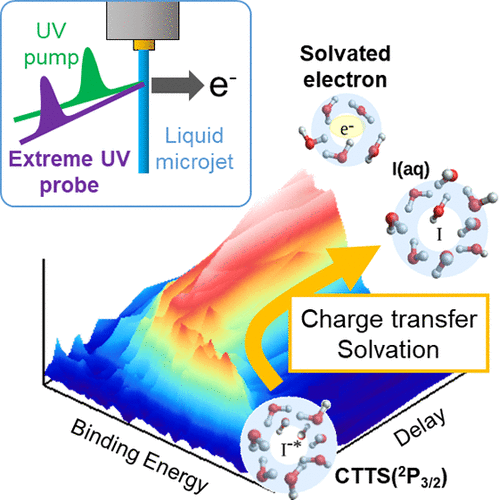
Charge transfer reactions from I- to solvent water, methanol, and ethanol were studied using extreme ultraviolet time-resolved photoelectron spectroscopy (EUV-TRPES). This technique eliminates spectral broadening, previously seen in UV-TRPES, caused by electron inelastic scattering in liquids, and enables clear observation of the temporal evolution of the spectral shape. The peak position, width, and intensity of the electron binding energy distribution indicate electron detachment and subsequent solvation and thermalization processes. Geminate recombination between detached electrons and iodine atoms is discussed using a diffusion equation and a global fitting analysis based on a kinetics model.
Shallow and deep trap states of solvated electrons in methanol and their formation, electronic excitation, and relaxation dynamics
Jinggang Lan, Yo-ichi Yamamoto, Toshinori Suzuki, and Vladimir Rybkin
Chem. Sci., 13, 3837−3844 (2022)
We present condensed-phase first-principles molecular dynamics simulations to elucidate the presence of different electron trapping sites in liquid methanol and their roles in the formation, electronic transitions, and relaxation of solvated electrons (emet−) in methanol. Excess electrons injected into liquid methanol are most likely trapped by methyl groups, but rapidly diffuse to more stable trapping sites with dangling OH bonds. After localization at the sites with one free OH bond (1OH trapping sites), reorientation of other methanol molecules increases the OH coordination number and the trap depth, and ultimately four OH bonds become coordinated with the excess electrons under thermal conditions. The simulation identified four distinct trapping states with different OH coordination numbers. The simulation results also revealed that electronic transitions of emet− are primarily due to charge transfer between electron trapping sites (cavities) formed by OH and methyl groups, and that these transitions differ from hydrogenic electronic transitions involving aqueous solvated electrons (eaq−). Such charge transfer also explains the alkyl-chain-length dependence of the photoabsorption peak wavelength and the excited-state lifetime of solvated electrons in primary alcohols.
Exploration of gas−liquid interfaces for liquid water and methanol using extreme ultraviolet laser photoemission spectroscopy
Yo-ichi Yamamoto, Tatsuya Ishiyama, Akihiro Morita, and Toshinori Suzuki
J. Phys. Chem. B, 125, 10514−10526 (2021)
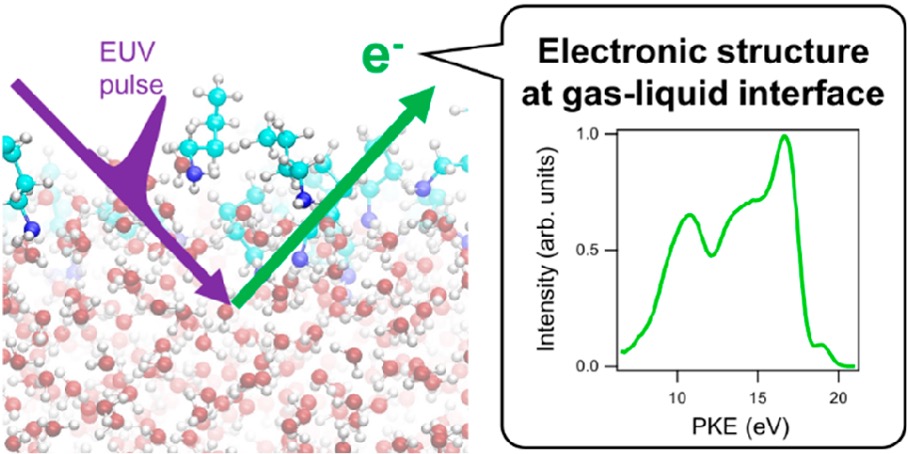
We present a study using extreme UV (EUV) photoemission spectroscopy of the valence electronic structures of aqueous and methanol solutions using a 10 kHz EUV light source based on high-order harmonic generation and a magnetic bottle time-of-flight electron spectrometer. Two aspects of the observed spectra are highlighted in this study. One is variation of the vertical ionization energy (VIE) for liquids as a function of the solute concentration, which is closely related to surface dipoles at the gas–liquid interface. The experimental results show that the VIE of liquid water increases slightly with increasing concentrations of NaCl and NaI and decreases with NaOH. The VIE of liquid methanol was also found to change slightly with NaI. On the other hand, tetrabutylammonium iodide (TBAI) and butylamine (BA) clearly reduce the VIE for liquid water, which is attributed to the formation of an electric double layer (EDL) by segregated solutes at the gas–liquid interface. As evidence for this, when the pH of an aqueous BA solution is reduced to protonate BA, the VIE shift gradually decreases because the protonated BA moves into the bulk to suppress the influence of the EDL. We computed the surface potentials for these solutions using molecular dynamics simulations, and the results supported our interpretation of the experimental results. Another observation is the variation of the relative energy and shape of individual photoelectron bands for solvents, which is related to alteration of the structure and constituents of the first solvation shell of ionized solvent molecules. All of the solutes cause changes in the photoelectron spectra at high concentration, one of the most prominent of which is the degree of splitting of the 3a1 band for liquid water and the 7a' band for liquid methanol, which are sensitive to hydrogen bonding in the liquids. The 3a1 splitting decreases with the increasing concentration of NaI, NaCl, and NaOH, indicating that Na+ penetrates into the hydrogen-bonding network to coordinate to a nonbonding electron of a water molecule. On the other hand, TBAI and BA cause smaller changes in the 3a1 splitting. Full interpretation of these spectroscopic features awaits extensive quantum chemical calculations and is beyond the scope of this study. However, these results illustrate the strong potential of EUV laser photoemission spectroscopy of liquids for exploration of interfacial and solution chemistry.
Design and characterization of a magnetic bottle electron spectrometer for time-resolved extreme UV and X-ray photoemission spectroscopy of liquid microjets
Naoya Kurahashi, Stephan Thürmer, Suet Yi Liu, Yo-ichi Yamamoto, Shutaro Karashima, Atanu Bhattacharya, Yoshihiro Ogi, Takuya Horio, and Toshinori Suzuki
Struct. Dyn., 8, 034303 (2021)
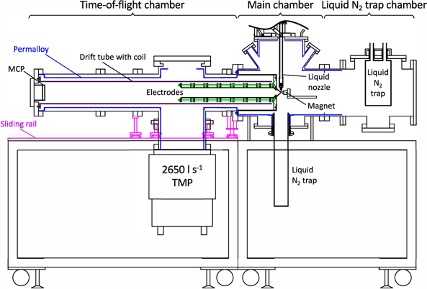
We describe a magnetic bottle time-of-flight electron spectrometer designed for time-resolved photoemission spectroscopy of a liquid micro- jet using extreme UV and X-ray radiation. The spectrometer can be easily reconfigured depending on experimental requirements and the energy range of interest. To improve the energy resolution at high electron kinetic energy, a retarding potential can be applied either via a stack of electrodes or retarding mesh grids, and a flight-tube extension can be attached to increase the flight time. A gated electron detector was developed to reject intense parasitic signal from light scattered off the surface of the cylindrically shaped liquid microjet. This detector features a two-stage multiplication with a microchannel plate plus a fast-response scintillator followed by an image-intensified photon detec- tor. The performance of the spectrometer was tested at SPring-8 and SACLA, and time-resolved photoelectron spectra were measured for an ultrafast charge transfer to solvent reaction in an aqueous NaI solution with a 200 nm UV pump pulses from a table-top ultrafast laser and the 5.5 keV hard X-ray probe pulses from SACLA.
Valence photoelectron spectra of liquid methanol and ethanol measured using HeII radiation
Stephan Thürmer, Takatoshi Shinno, and Toshinori Suzuki
J. Phys. Chem. A, 125, 2492-2503 (2021)
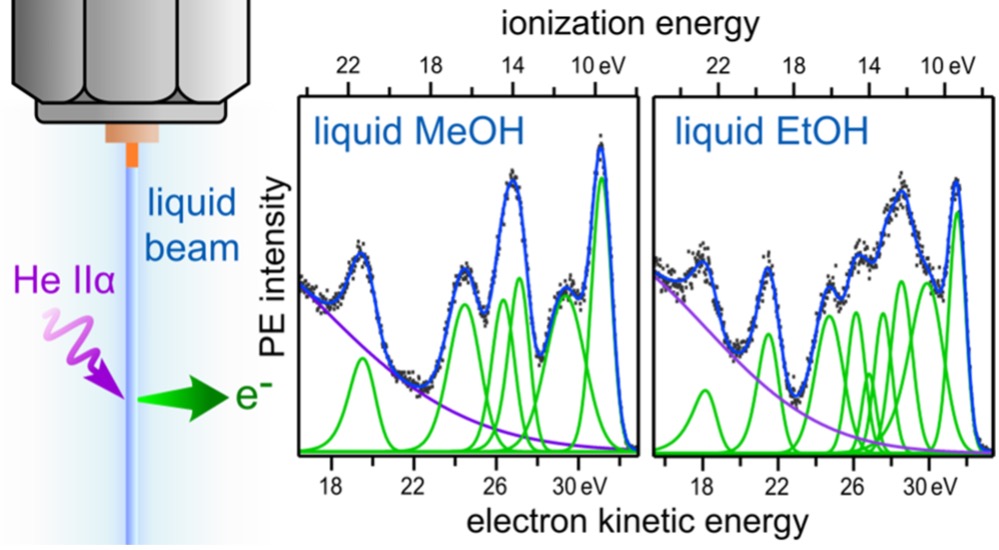
High-resolution photoelectron (PE) spectra of liquid methanol and ethanol were measured using a liquid microjet and He IIα radiation (40.813 eV). The vertical ionization energy and the ionization threshold were determined as 9.70 ± 0.07 and 8.69 ± 0.07 eV for methanol and 9.52 ± 0.07 and 8.52 ± 0.07 eV for ethanol, respectively. Individual photoemission bands observed for the liquids are well correlated with those in PE spectra of the gaseous samples also measured in the present study, except that the liquid band positions were shifted on average by −1.23 eV for methanol and −1.10 eV for ethanol as compared to the gas. The 5a' and 7a' bands of liquid methanol exhibit specifically larger broadening than other bands, for which we attempted spectral fitting with two components, similarly with the case of the 3a1 band of liquid water. PE spectra of both liquid and gaseous ethanol are congested partly due to the presence of the trans and gauche isomers; however, the overall band positions are generally in good agreement with predictions based on quantum chemical calculations. Comparison of the measured PE spectra with experimental and simulated X-ray emission spectra indicate that spectral differences in the lowest ionization band of both methanol and ethanol originate from involvement of nuclear dynamics in the X-ray emission process.
Ultrafast dynamics of water radiolysis: hydrated electron formation, solvation, recombination, and scavenging
Yo-ichi Yamamoto and Toshinori Suzuki
J. Phys. Chem. Lett., 11, 5510-5516 (2020)
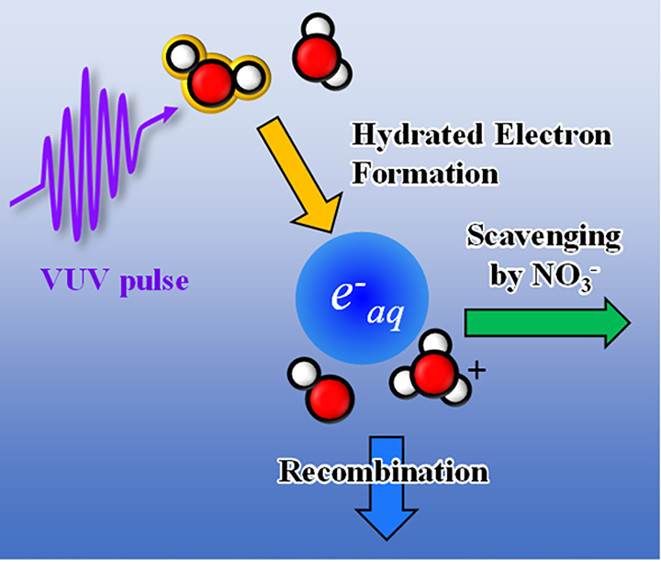
The ultrafast formation, solvation, and geminate recombination of hydrated electrons upon vacuum ultraviolet photoexcitation of liquid water and the static and dynamic scavenging by NO3– are investigated using femtosecond time-resolved photoelectron spectroscopy. The solvation time constant for excess electrons is typical of that for liquid water but increases slightly with increasing excitation energy. The electron survival probability for geminate recombination is found to be much lower than the literature values owing to previously unobserved ultrafast geminate recombination in a period of 5 ps. NO3– induces the ultrafast (static) scavenging of photoexcited electronic states of liquid water and the dynamic scavenging of detached electrons with a reaction rate that is dependent on the excitation energy. The formation of hydrated electrons at 7.7 eV is ascribed to a H-atom-transfer process, but it is plausible that additional formation channels open at higher energies.
Surface potential of liquid microjet investigated using extreme ultraviolet photoelectron spectroscopy
Junichi Nishitani, Shutaro Karashima, Christopher W. West, and Toshinori Suzuki
J. Chem. Phys., 152, 144503 (2020)
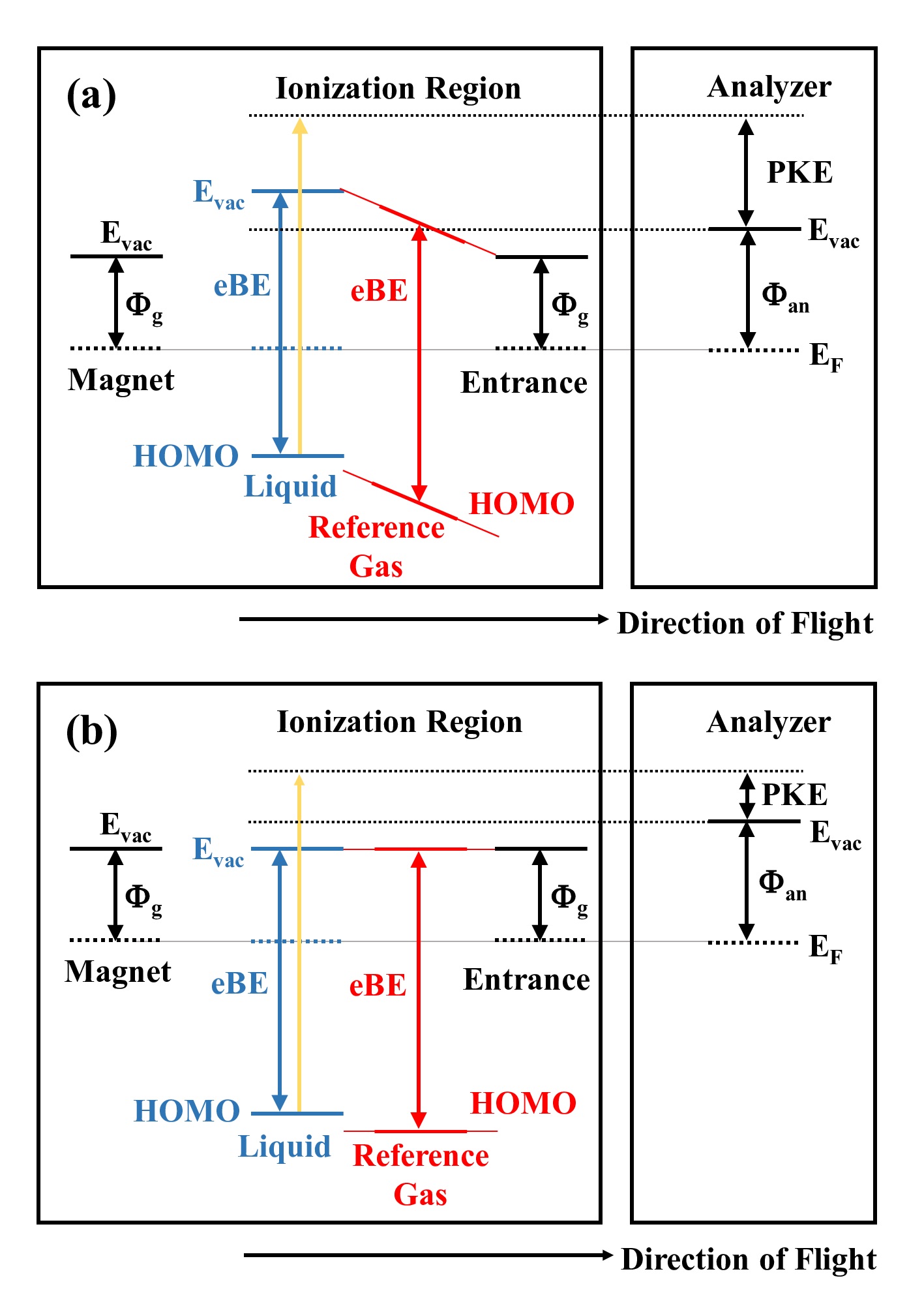
Photoelectron spectroscopy of a liquid microjet requires careful energy calibration against electrokinetic charging of the microjet. For minimizing the error from this calibration procedure, Kurahashi et al. previously suggested optimization of an electrolyte concentration in aqueous solutions [Kurahashi et al., J. Chem. Phys. 140, 174506 (2014)]. More recently, Olivieri et al. proposed an alternative method of applying a variable external voltage on the liquid microjet [Olivieri et al., Phys. Chem. Chem. Phys. 18, 29506 (2016)]. In this study, we examined these two methods of calibration using extreme ultraviolet photoelectron spectroscopy with a magnetic bottle time-of-flight photoelectron spectrometer. We confirmed that the latter method flattens the vacuum level potential around the microjet, similar to the former method, while we found that the applied voltage energy-shifts the entire spectrum. Thus, careful energy recalibration is indispensable after the application of an external voltage for accurate measurements. It is also pointed out that electric conductivity of liquid on the order of 1 mS/cm is required for stable application of an external voltage. Therefore, both methods need a similar concentration of an electrolyte. Using the calibration method proposed by Olivieri et al., Perry et al. have recently revised the vertical ionization energy of liquid water to be 11.67(15) eV [Perry et al., J. Phys. Chem. Lett. 11, 1789 (2020)], which is 0.4 eV higher than the previously estimated value. While the source of this discrepancy is still unclear, we estimate that their calibration method possibly leaves uncertainty on the order of 0.1 eV.
Extreme ultraviolet time-resolved photoelectron spectroscopy of aqueous aniline solution: enhanced surface concentration and pump-induced space charge effect
Christopher W. West, Junichi Nishitani, Chika Higashimura, and Toshinori Suzuki
Mol. Phys., 119, e1748240 (2020)
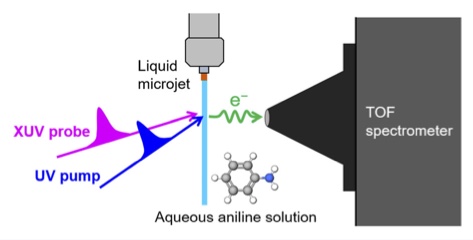
We present extreme ultraviolet (XUV) time-resolved photoelectron spectroscopy (TRPES) of an aqueous aniline solution. One-colour XUV-induced photoemission signal of aniline was observed with much greater intensity than expected from its molar fraction in the bulk solution, indicating that aniline is hydrophobically segregated on the liquid surface. The concentration dependence of the photoelectron intensity is found to be well correlated with the surface concentration of aniline estimated by surface tension measurements. Similar segregation was observed also for phenol in aqueous solution. The enhanced surface concentration of aniline makes its XUV-TRPES to be highly vulnerable to the pump-induced space charge effects (PISC). The PISC caused by a moderate pump intensity was not completely corrected using a simple mean field model, and reduction of the pump pulse intensity was necessary. The spectra measured at lower pump intensity were corrected for PISC, which enabled us to extract the information on the excited state dynamics of aniline in aqueous solution under 240 nm photoexcitation. Two components with lifetime on sub-ps and > 100 ps timescales were determined, and the former is ascribed to the solvation dynamics in the S1 state after the ultrafast internal conversion from the S3 state and the latter to the subsequent population decay of the S1 state.
Solvated electron formation from the conduction band of liquid methanol: Transformation from a shallow to deep trap state
Ayano Hara, Yo-ichi Yamamoto, and Toshinori Suzuki
J. Chem. Phys, 151, 114503 (2019)
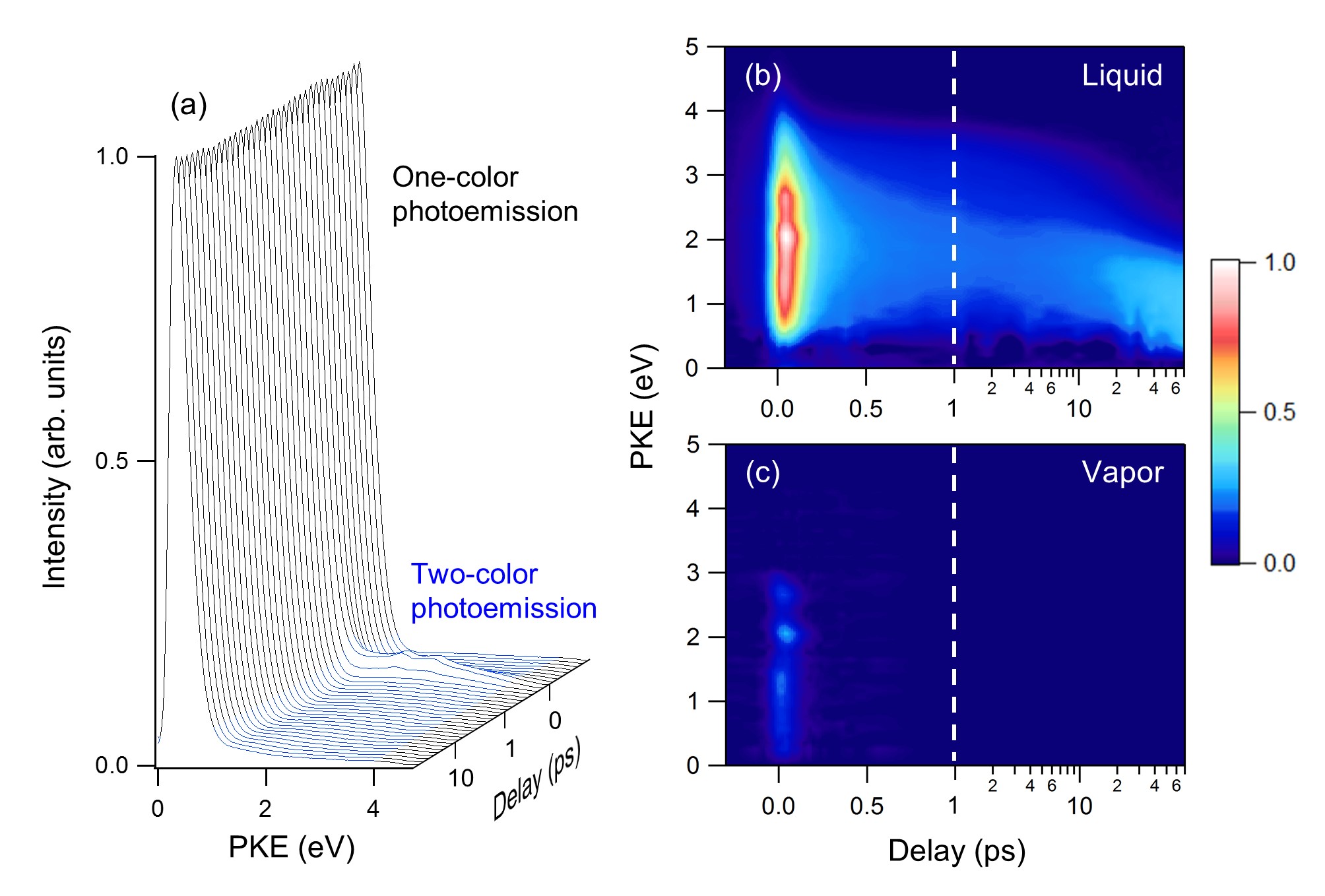
We report solvated electron (e−solv) formation dynamics from the conduction band of liquid methanol studied using femtosecond time-resolved photoelectron spectroscopy. Liquid methanol is excited with vacuum UV (9.3 eV) pump pulses, and the subsequent electron dynamics are probed with UV pulses. The photoelectron signal exhibits a short-lived component (τ = 85 fs) without spectral evolution followed by a long-lived component with continuous spectral evolution over tens of picoseconds. We ascribe the former to a superexcited state, most likely the Wannier exciton, and the latter to the ground electronic state of e−solv. In order to extract accurate energetics from the observed photoelectron spectra, we employ a spectral retrieval method to account for spectral broadening and shifting due to inelastic scattering of photoelectrons in the liquid. The electron binding energy (eBE) of the initial trap state of an electron is determined to be about 1.5 eV, and its biexponential increase up to 3.4 eV is observed with time constants of 2 and 31 ps, which are greater than 0.27 and 13 ps observed for e−solv created by the charge-transfer-to-solvent reaction from CH3O− to liquid methanol. The solvation dynamics of e−solv created by the electron trapping exhibit a pseudoisosbestic point at a pump-probe delay time of around 15 ps, and the peak energy of the eBE distribution rapidly changes around that time. These results indicate that there exist two trap states, both of which exhibit increasing eBE with time; however, the eBE of the shallow trap state increases only up to 2.1 eV, and transformation to a deep trap state at 25 ps occurs to reach an eBE of 3.4 eV.
Binding energy of solvated electrons and retrieval of true UV photoelectron spectra of liquids
Junichi Nishitani, Yo-ichi Yamamoto, Christopher W. West, Shutaro Karashima, and Toshinori Suzuki
Sci. Adv., 5, eaaw6896 (2019)
The electronic energy and dynamics of solvated electrons, the simplest yet elusive chemical species, is of interest in chemistry, physics, and biology. Here, we present the electron binding energy distributions of solvated electrons in liquid water, methanol, and ethanol accurately measured using extreme ultraviolet (EUV) photoelectron spectroscopy of liquids with a single-order high harmonic. The distributions are Gaussian in all cases. Using the EUV and UV photoelectron spectra of solvated electrons, we succeeded in retrieving sharp electron kinetic energy distributions from the spectra broadened and energy shifted by inelastic scattering in liquids, overcoming an obstacle in ultrafast UV photoelectron spectroscopy of liquids. The method is demonstrated for the benchmark systems of charge transfer to solvent reaction and ultrafast internal conversion of hydrated electron from the first excited state.
Ultrafast internal conversion and solvation of electrons in water, methanol and ethanol
Shutaro Karashima, Yo-ichi Yamamoto, and Toshinori Suzuki
J. Phys. Chem. Lett., 10, 4499-4504 (2019)
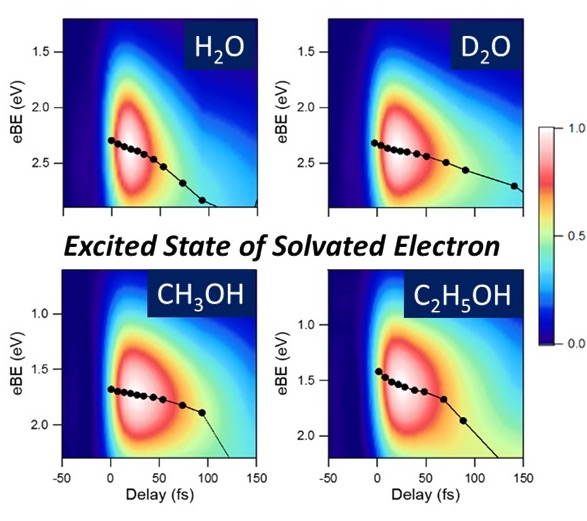
Ultrafast internal conversion from the first excited state of a solvated electron in water, methanol, and ethanol is investigated using time-resolved photoelectron spectroscopy of liquid microjets and a spectral retrieval method. Photoelectron spectra corrected for inelastic scattering clearly reveal well-separated signals from the excited and ground states, and the latter enables us to analyze the solvation dynamics in the ground state after internal conversion. Measurements with 25 fs time resolution identify a rapid increase in the vertical electron binding energy of the solvated electron owing to nuclear wave packet motions in the excited state and allow us to precisely determine the internal conversion time.
Charge-transfer-to-solvent reaction in a hydrophobic tetrabutylammonium iodide molecular layer in aqueous solution
Shutaro Karashima and Toshinori Suzuki
J. Phys. Chem. B, 123, 3769-3775 (2019)
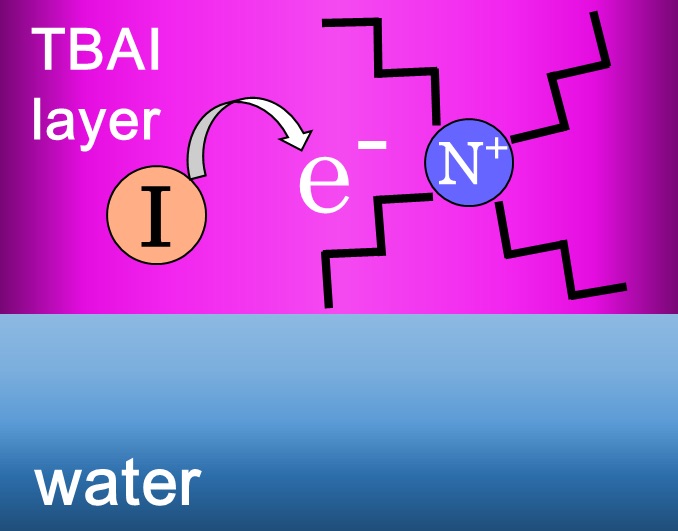
We present ultrafast photoelectron spectroscopy of the charge-transfer-to-solvent reaction in a segregated TBAI (tetrabutylammonium iodide) molecular layer in aqueous solution. The reaction times and electron binding energies of transient species vary with TBAI concentration from a very low value of 1 × 10-3 mol L-1, which is in contrast to NaI solution exhibiting no concentration (0.01−1.0 mol L-1) dependence. The result from soft X-ray N(1s) spectroscopy indicates that the photoelectron intensity in TBAI aqueous solution is about 70 times enhanced as compared to that in NH4Cl aqueous solution for an identical salt concentration, and TBA+ drags I- to the surface region. At high TBAI concentrations, electrons released from I- are trapped and held in the TBAI molecular layer owing to electrostatic attraction by TBA+.
On the relation between the interfacial charge of a discharging nozzle and electrification of a liquid microjet
Naoya Kurahashi and Toshinori Suzuki
Chem. Lett., 47, 1, 16-19 (2018)
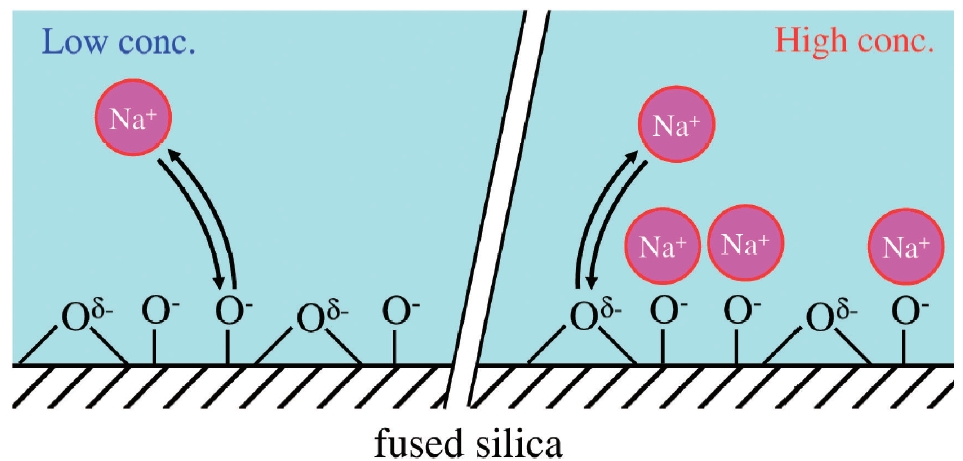
A liquid microjet discharged from a fused silica capillary is electrified, and the polarity of thiselectrification is opposite to that of the net charge at the solid-liquid interface of the capillary nozzle.
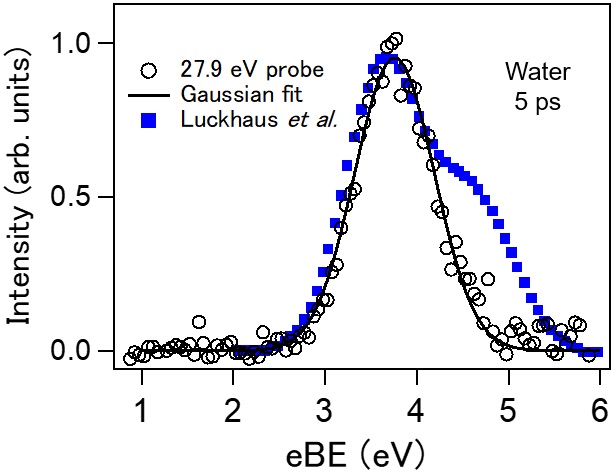
Genuine binding energy of the hydrated electron
David Luckhaus, Yo-ichi Yamamoto, Toshinori Suzuki, and Ruth Signorell
Sci. Adv., 3, e1603224 (2017)
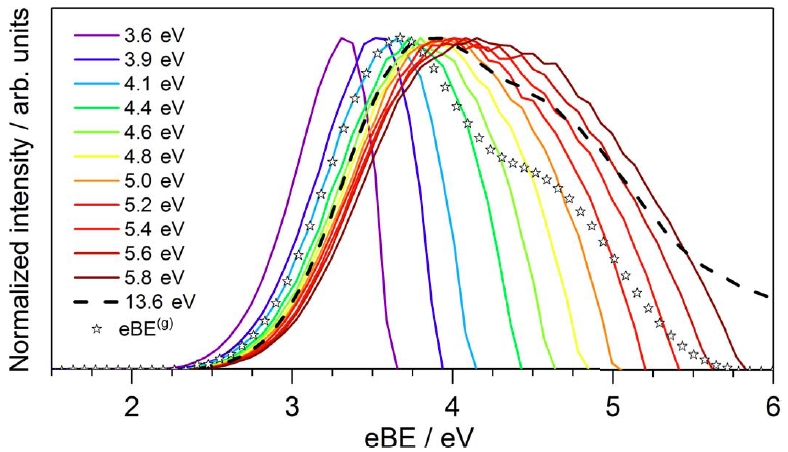
The unknown influence of inelastic and elastic scattering of slow electrons in water has made it difficult to clarify the role of the solvated electron in radiation chemistry and biology. We combine accurate scattering simulations with experimental photoemission spectroscopy of the hydrated electron in a liquid water microjet, with the aim of resolving ambiguities regarding the influence of electron scattering on binding energy spectra, photoelectron angular distributions, and probing depths. The scattering parameters used in the simulations are retrieved from independent photoemission experiments of water droplets. For the ground-state hydrated electron, we report genuine values devoid of scattering contributions for the vertical binding energy and the anisotropy parameter of 3.7 ± 0.1 eV and 0.6 ± 0.2, respectively. Our probing depths suggest that even vacuum ultraviolet probing is not particularly surface-selective. Our work demonstrates the importance of quantitative scattering simulations for a detailed analysis of key properties of the hydrated electron.
Angle-resolved photoemission spectroscopy of liquid water at 29.5 eV
Junichi Nishitani, Christopher W. West, and Toshinori Suzuki
Struct. Dyn., 4, 044014 (2017)
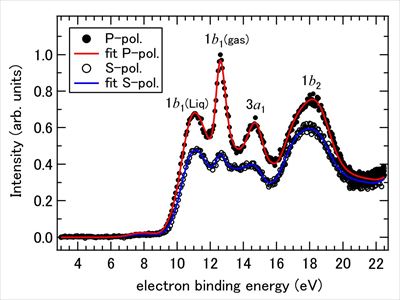
Angle-resolved photoemission spectroscopy of liquid water was performed using extreme ultraviolet radiation at 29.5 eV and a time-of-flight photoelectron spectrometer. SiC/Mg coated mirrors were employed to select the single-order 19th harmonic from laser high harmonics, which provided a constant photon flux for different laser polarizations. The instrument was tested by measuring photoemission anisotropy for rare gases and water molecules and applied to a microjet of an aqueous NaI solution. The solute concentration was adjusted to eliminate an electric field gradient around the microjet. The observed photoelectron spectra were analyzed considering contributions from liquid water, water vapor, and an isotropic background. The anisotropy parameters of the valence bands (1b1, 3a1, and 1b2) of liquid water are considerably smaller than those of gaseous water, which is primarily attributed to electron scattering in liquid water.
Charge-transfer-to-solvent reactions from I- to water, methanol, and ethanol studied by time-resolved photoelectron spectroscopy of liquids
Haruki Okuyama, Yoshi-Ichi Suzuki, Shutaro Karashima, and Toshinori Suzuki
J. Chem. Phys., 145, 074502 (2016)
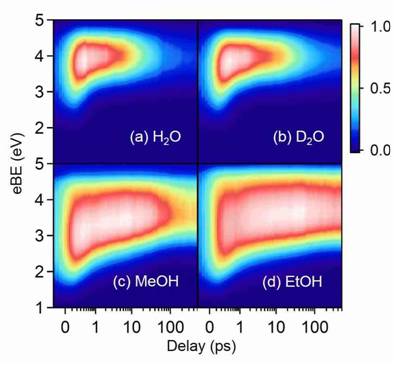
The charge-transfer-to-solvent (CTTS) reactions from iodide (I-) to H2O, D2O, methanol, and ethanol were studied by time-resolved photoelectron spectroscopy of liquid microjets using a magnetic bottle time-of-flight spectrometer with variable pass energy. Photoexcited iodide dissociates into a weak complex (a contact pair) of a solvated electron and an iodine atom in similar reaction times, 0.3 ps in H2O and D2O and 0.5 ps in methanol and ethanol, which are much shorter than their dielectric relaxation times. The results indicate that solvated electrons are formed with minimal solvent reorganization in the long-range solvent polarization field created for I-. The photoelectron spectra for CTTS in H2O and D2O measured with higher accuracy than in our previous study [Chem. Sci. 2 1094 (2011)] indicate that internal conversion yields from the photoexcited I-* (CTTS) state are less than 10%, while alcohols provide 2 - 3 times greater yields of internal conversion from I-*. The overall geminate recombination yields are found to be in the order of H2O > D2O > methanol > ethanol, which is opposite to the order of the mutual diffusion rates of an iodine atom and a solvated electron. This result is consistent with transition state theory for an adiabatic outer-sphere electron transfer process, which predicts that the recombination reaction rate has a pre-exponential factor inversely proportional to a longitudinal solvent relaxation time.
Wavelength Dependence of UV Photoemission from Solvated Electrons in Bulk Water, Methanol, and Ethanol
Yo-Ichi Yamamoto, Shutaro Karashima, Shunsuke Adachi, and Toshinori Suzuki
J. Phys. Chem. A, 120, 1153 - 1159 (2016)
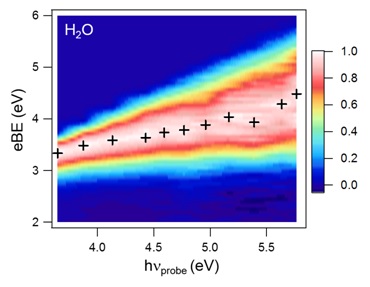
We have measured the wavelength dependence (340 - 215 nm) of one-photon photoemission from the ground electronic state of solvated electrons in bulk water, methanol, and ethanol. In every case, the vertical electron binding energy (VBE) gradually increased with photon energy, indicating that the photoelectron kinetic energy diminishes as a result of electron-vibration inelastic scattering prior to emission from the liquid surface. In contrast, the VBE of the Rydberg electron in DABCO (1,4-diazabicyclo[2,2,2]octane), which has a surface-excess density, revealed no clear wavelength dependence. These results suggest that the solvated electrons are created predominantly in the bulk and that VBEs measured using UV photoemission spectroscopy of liquids generally require energy corrections to account for inelastic scattering effects. From the wavelength dependence, we have re-estimated the VBEs of solvated electrons in bulk water, methanol, and ethanol to be 3.3, 3.1, and 3.1 eV, respectively. Hydrated electrons were also identified by photoemission spectroscopy using 90 nm radiation.
Resolving non-adiabatic dynamics of hydrated electrons using ultrafast photoemission anisotropy
Shutaro Karashima, Yo-ichi Yamamoto, and Toshinori Suzuki
Phys. Rev. Lett., 116, 137601 (2016)
We have studied ultrafast non-adiabatic dynamics of excess electrons trapped in the band gap of liquid water using time- and angle-resolved photoemission spectroscopy. Anisotropic photoemission from the first excited state was discovered, which enabled unambiguous identification of non-adiabatic transition to the ground state in 60 fs in H2O and 100 fs in D2O. The photoelectron kinetic energy distribution exhibited rapid spectral shift in ca. 20 fs, which is ascribed to librational response of a hydration shell to electronic excitation. Photoemission anisotropy indicates that the electron orbital in the excited state is depolarized in less than 40 fs.
Effective attenuation length of an electron in liquid water between 10 and 600 eV
Yoshi-Ichi Suzuki, Kiyoshi Nishizawa, Naoya Kurahashi, and Toshinori Suzuki
Phys. Rev. E, 90, 010302(R) (2014)
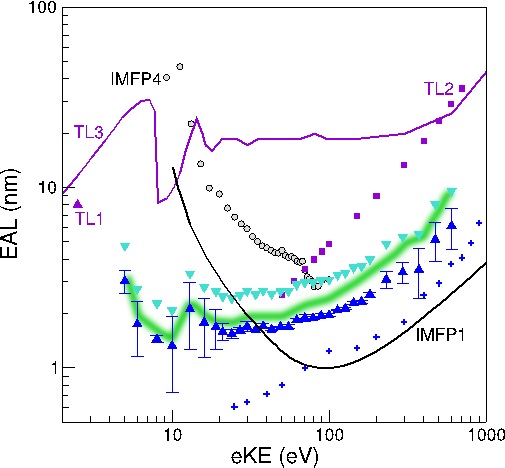
The absolute values of the effective attenuation length of an electron in liquid water are determined using soft x-ray O1s photoemission spectroscopy of a liquid beam of water without employing any theoretical estimation or computationally obtained value. The effective attenuation length is greater than 1 nm in the entire electron kinetic energy region and exhibits very flat energy dependence in the 10-100 eV region.
Time- and Angle-Resolved Photoemission Spectroscopy of Hydrated Electrons Near a Liquid Water Surface
Yo-ichi Yamamoto, Yoshi-Ichi Suzuki, Gaia Tomasello, Takuya Horio, Shutaro Karashima, Roland Mitric, and Toshinori Suzuki
Phys. Rev. Lett., 112, 187603 (2014)
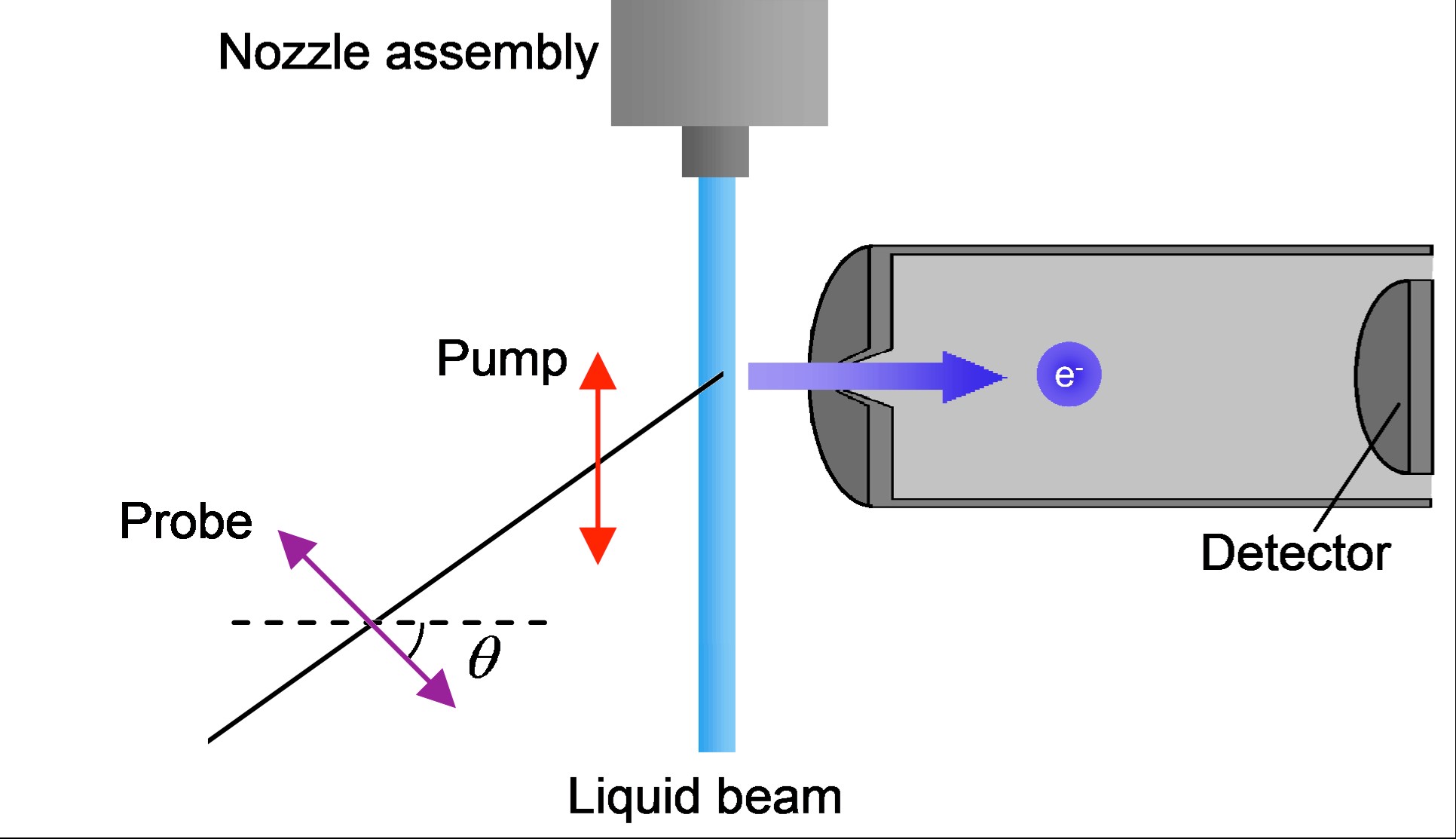
We present time- and angle-resolved photoemission spectroscopy of trapped electrons near liquid surfaces. Photoemission from the ground state of a hydrated electron at 260 nm is found to be isotropic, while anisotropic photoemission is observed for the excited states of 1,4-diazabicyclo[2,2,2]octane and I- in aqueous solutions. Our results indicate that surface and subsurface species create hydrated electrons in the bulk side. No signature of a surface-bound electron has been observed.
Photoelectron spectroscopy of aqueous solutions: Streaming potentials of NaX (X = Cl, Br, and I) solutions and electron binding energies of liquid water and X-
Naoya Kurahashi, Shutaro Karashima, Ying Tang, Takuya Horio, Bumaliya Abulimiti, Yoshi-Ichi Suzuki, Yoshihiro Ogi, Masaki Oura, and Toshinori Suzuki
J. Chem. Phys., 140, 174506 (2014)
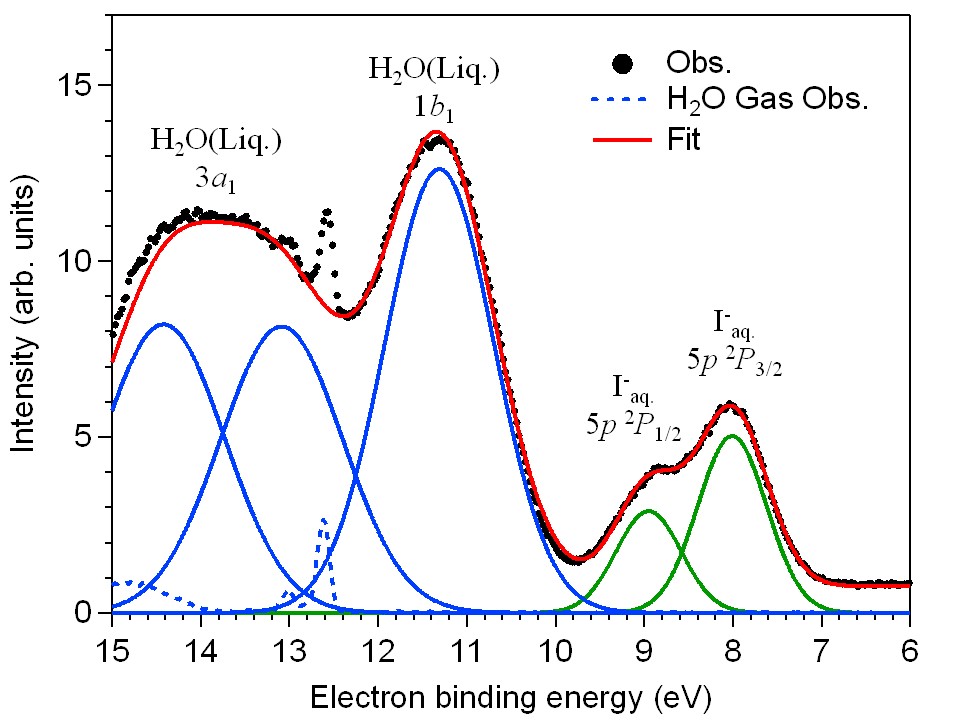
The streaming potentials of liquid beams of aqueous NaCl, NaBr, and NaI solutions are measured using soft X-ray, He(I), and laser multiphoton ionization photoelectron spectroscopy. Gaseous molecules are ionized in the vicinity of liquid beams and the photoelectron energy shifts are measured as a function of the distance between the ionization point and the liquid beam. The streaming potentials change their polarity with concentration of electrolytes, from which the singular points of concentration eliminating the streaming potentials are determined. The streaming currents measured in air also vanish at these concentrations. The electron binding energies of liquid water and I-, Br-, and Cl- anions are revisited and determined more accurately than in previous studies.
Photoelectron spectra of solvated electrons in bulk water, methanol, and ethanol
Takuya Horio, Huan Shen, Shunsuke Adachi, and Toshinori Suzuki
Chem. Phys. Lett., 535, 12-16 (2012)
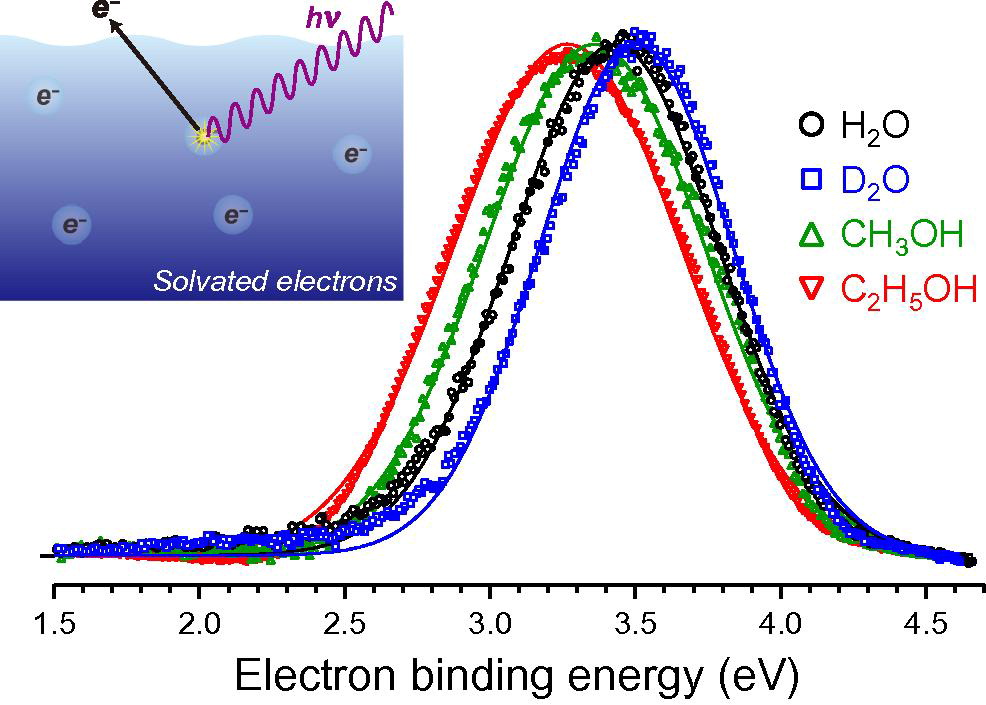
Photoelectron spectra of solvated electrons in bulk liquids were obtained at energy-resolution of 60 meV using a linear time-of-flight photoelectron spectrometer and a 100 kHz ultraviolet femtosecond laser. Solvated electrons in H2O, D2O, methanol, and ethanol were generated by 226 nm excitation of the charge-transfer-to-solvent bands of I- in 0.1 M NaI solutions, and the photoelectron spectra were measured using 260 nm pulses with a time delay of 2 ns. The electron binding energies and band shapes are discussed.
Isotope effect on ultrafast charge-transfer-to-solvent reaction from I- to water in aqueous NaI solution
Yoshi-Ichi Suzuki, Huan Shen, Ying Tang, Naoya Kurahashi, Kentaro Sekiguchi, Tomoya Mizuno, and Toshinori Suzuki
Chemical Science, 2 (6), 1094-1102 (2011)
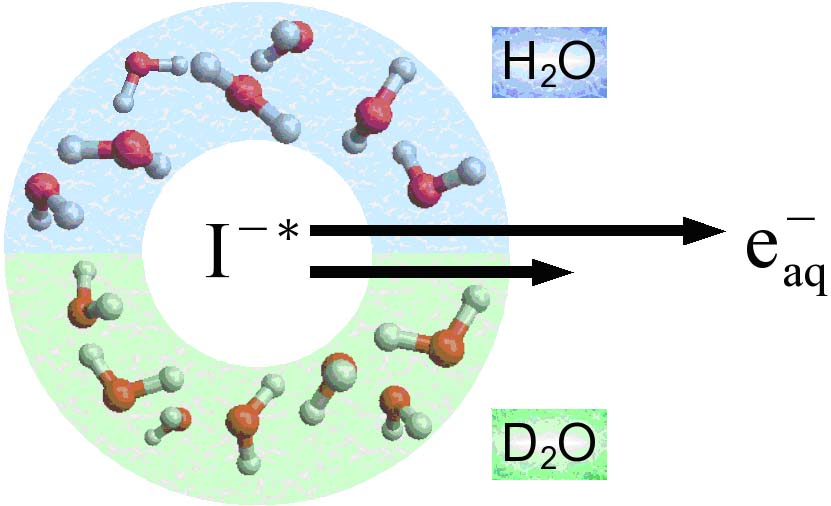
A charge-transfer-to-solvent (CTTS) reaction from a photoexcited iodine atomic anion, I-(aq), in bulk water (H2O and D2O) was studied by time-resolved photoelectron spectroscopy using a liquid beam (microjet) of aqueous NaI solution. The 2P3/2 CTTS state of I-(aq) was excited by a 226 nm femtosecond laser pulse and the evolution of the nonstationary electronic state was probed using another ultraviolet femtosecond laser pulse. Global fitting of the observed time-dependent photoelectron kinetic energy distributions provided the time constants of individual reaction steps and the photoelectron spectra from the CTTS state, a contact pair, the solvent-separated state, and a hydrated electron. Most of the elementary reaction steps revealed a strong deuterium isotope effect, indicating coupling of the electron dynamics and the hydrogen atomic motion of solvent water. However, nondiffusive geminate recombination processes from the CTTS state and a contact pair were almost insensitive to deuteration. Consequently, geminate recombination processes from the CTTS state and a contact pair occurs more efficiently in D2O, because the response of water is decelerated in D2O. In contrast, the recombination process from the solvent-separated state in the final step of the CTTS reaction is less efficient in D2O, presumably due to the smaller zero point energy.
High-resolution soft X-Ray photoelectron spectroscopy of liquid water
Kiyoshi Nishizawa, Naoya Kurahashi, Kentarou Sekiguchi, Tomoya Mizuno, Yoshihiro Ogi, Takuya Horio, Masaki Oura, Nobuhiro Kosugi, and Toshinori Suzuki
Phys. Chem. Chem. Phys., 13 (2), 413-417 (2011)
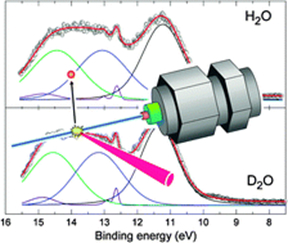
High-resolution soft X-ray photoelectron spectra of liquid water (H2O and D2O) were measured using a liquid beam photoelectron spectrometer. The 1a1 (O1s) band and the lowest valence 1b1 band had single peaks, which is not consistent with the split 1b1 → 1a1 of the X-ray emission band of liquid water if the splitting is assumed to originate from level shifts in two different hydrogen bonding structures. The second valence 3a1 band of liquid water exhibited a flat top implying that two bands exist underneath a broad feature, which is similar to the case of the 3a1 band of amorphous ice. The energy splitting between the two 3a1 bands is estimated to be 1.38 eV (H2O) and 1.39 eV (D2O). Ab initio calculations suggest that the large splitting of the 3a1 band is characteristic of water molecules that function as both proton donor and acceptor. The overall result is consistent with the conventional model of a tetrahedral hydrogen-bonding network in liquid water.
Time-resolved photoelectron spectroscopy of bulk liquids at ultra-low kinetic energy
Ying Tang, Yoshi-ichi Suzuki, Huan Shen, Kentaro Sekiguchia, Naoya Kurahashi, Kiyoshi Nishizawa, Peng Zuo, and Toshinori Suzuki
Chem. Phys. Lett., 494, 111-116 (2010)
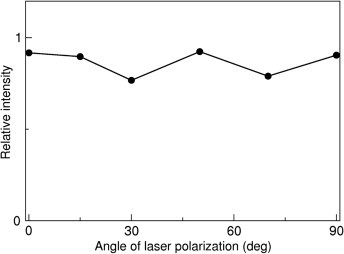
Time-resolved photoelectron spectroscopy (TR-PES) of ultrafast dynamics in solution is presented. To measure the photoelectron kinetic energy distribution (PKED) that is free from inelastic scattering in solution, photoelectrons were generated with ultra-low kinetic energies (ULKE: <5 eV). Time constants of the elementary processes in the charge-transfer-to-solvent (CTTS) reaction from I- to bulk water were in excellent agreement with those obtained by transient absorption spectroscopy, demonstrating the bulk-sensitivity of TR-PES-ULKE. The analysis suggests that the CTTS reaction proceeds via two intermediates, and that 30% of the first intermediate and 70% of the second intermediate respectively are quenched by geminate recombination between the electron and the neutral iodine atom.
Direct Measurement of Vertical Electron Binding Energies of Solvated Electrons in Methanol and Ethanol
Huan Shen, Naoya Kurahashi, Takuya Horio, Kentaro Sekiguchi, and Toshinori Suzuki
Chem. Lett., 39, 668-670 (2010)
Editor's choice
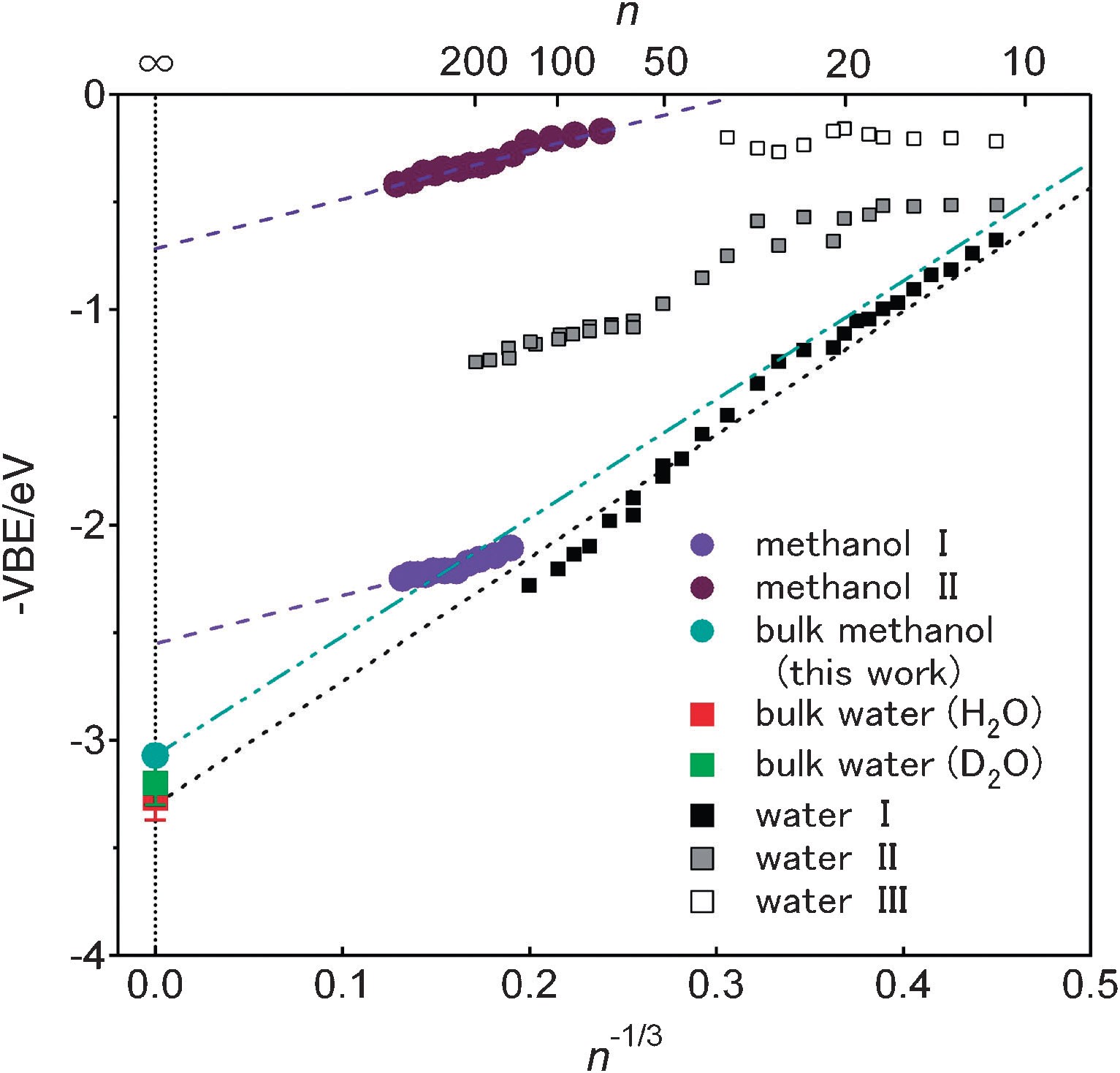
Time-resolved photoelectron spectroscopy at ultralow kinetic energy was applied to liquid beams of NaI solutions in methanol and ethanol. Solvated electrons were formed from I- ions in these solutions by charge transfer to solvent reactions. The vertical electron binding energies of the solvated electrons were determined for the first time. Both of the binding energies were found to be 3.10 ± 0.1 eV. This is in reasonable agreement with the dielectric continuum model of solvated electrons using the previously measured vertical binding energy of a hydrated electron. This indicates that the cavity radii of solvated electrons in water, methanol, and ethanol are approximately the same (0.33 - 0.35 nm).
Direct measurement of vertical binding energy of a hydrated electron
Ying Tang, Huan Shen, Kentaro Sekiguchi, Naoya Kurahashi, Yoshi-Ichi Suzuki, and Toshinori Suzuki
Phys. Chem. Chem. Phys., 12, 3653-3655 (2010)
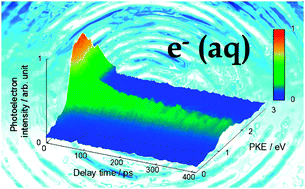
We present the first measurement of the vertical binding energy (VBE) of a hydrated electron in bulk water by the time-resolved photoelectron spectroscopy (TRPES) of the charge-transfer-to-solvent (CTTS) reaction in aqueous NaI solution. Our best estimate of VBE is 3.27 ± 0.10 eV for H2O and 3.20 ± 0.10 eV for D2O.
2. Ultrafast Photoelectron Spectroscopy of Isolated Molecules
Evidence for a Twisted Intermediate in Gaseous Uracil Revealed by Photoelectron Quantum Beats
Shutaro Karashima and Toshinori Suzuki
J. Phys. Chem. Lett., 16, 12045-12049 (2025)
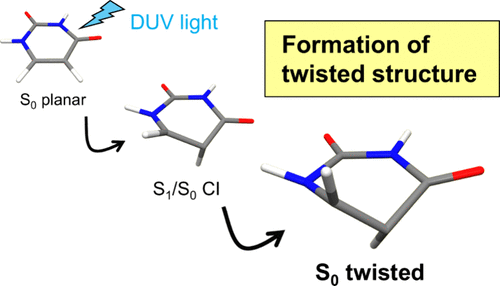
Recent theoretical and experimental investigations have identified the formation of a previously unrecognized intermediate characterized by a strongly twisted C5=C6 double bond in the ground electronic state of pyrimidine nucleobases in an aqueous solution. This intermediate is produced by ultrafast internal conversion, representing a critical pathway in the photophysics of nucleobase relaxation. Nevertheless, the existence of such a species under gas-phase conditions has not yet been confirmed. In this study, we present the first direct experimental evidence for the formation of twisted intermediates in isolated molecules. Analysis of vibrational quantum beats observed using extreme ultraviolet time-resolved photoelectron spectroscopy reveals the presence of this transient species in the gas phase, further confirming that its formation is an intrinsic property of the nucleobase chromophore.
Effects of Methyl Substitution on the Ultrafast Internal Conversion of Benzene
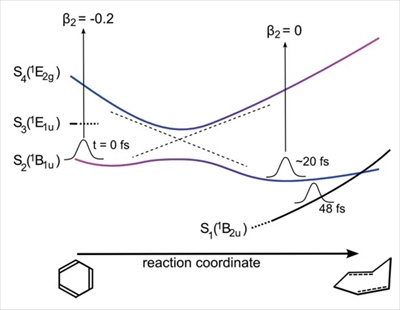
Kazuki Maeda, Alexie Boyer, Shutaro Karasshima, Alexander Humeniuk, and Toshinori Suzuki
J. Phys. Chem. Lett., 15, 11760-11766 (2024)
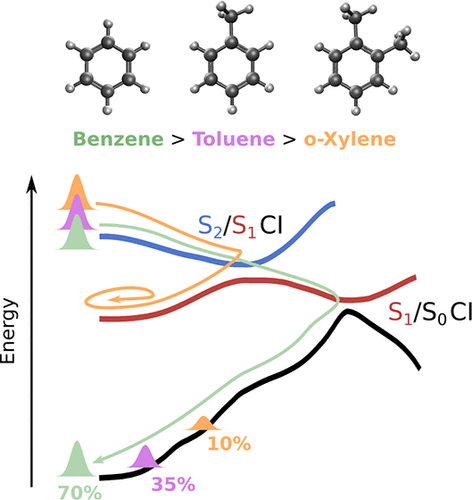
The effects of methyl substitution on the ultrafast internal conversion from the S2(1B1u, ππ*) state to the S0 state of benzene were studied using ultrafast extreme-ultraviolet photoelectron spectroscopy and electronic structure calculations. The quantum yield of the internal conversion to the S0 state reached ∼0.69 in benzene, while lower values of 0.35 and 0.12 were obtained for toluene and o-xylene, respectively. These results indicate that methyl substitution makes the conical intersections less accessible to the nuclear wave packet.
Deuterium Isotope Effect on Internal Conversion of Ethylene Studied by Time-Resolved Photoelectron Spectroscopy
Alexie Boyer, Alexander Humeniuk, Shutaro Karasshima, and Toshinori Suzuki
J. Phys. Chem. A, 128, 7068-7072 (2024)
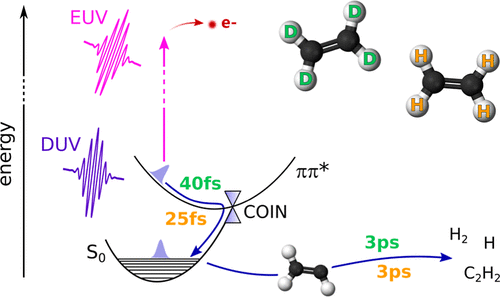
The effect of deuterium isotopes on the internal conversion of ethylene is studied by using extreme ultraviolet time-resolved photoelectron spectroscopy. For deuterium-labeled ethylene, the time scale for ultrafast internal conversion is increased by a factor of approximately √2, in agreement with the results of ab initio multiple spawning calculations, indicating the essential role played by hydrogen motion in the conversion process. Following internal conversion, a metastable species with an electron binding energy of ∼9 eV is produced, and it decays with a time constant similar to that for both isotopologues.
Vibrational Motions in Ultrafast Electronic Relaxation of Pyrazine
Shutaro Karashima, Alexander Humeniuk, and Toshinori Suzuki
J. Am. Chem. Soc., 146, 11067-11071 (2024)
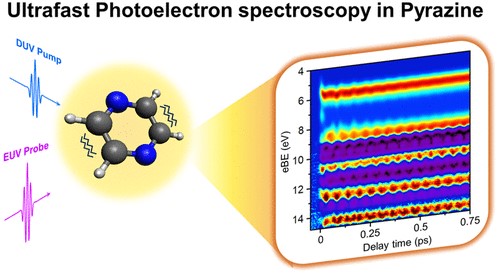
Ultrafast internal conversion via a conical intersection is ubiquitous in highly efficient photochemical reactions. Internal conversion from the 1ππ* to the 1nπ* state of pyrazine is the paradigm for this phenomenon; however, the relaxation occurs in such a short time (<20 fs) that the nuclear motion is difficult to observe in real time. The present study precisely measures the vibrational coherence transferred from the 1ππ* state to the 1nπ* state using time-resolved photoelectron spectroscopy with an unprecedented time resolution of 13.3 fs and reveals the key nuclear motions that drive the internal conversion.
Singnatures of Conical Intersections in Extreme Ultrviolet Photoelectron Spectra of Furan Measured with 15 fs Time Resolution
Ryuta Uenishi, Alexie Boyer, Shutaro Karashima, Alexander Humeniuk, and Toshinori Suzuki
J. Phys. Chem. Lett., 15, 8, 2222-2227 (2024)
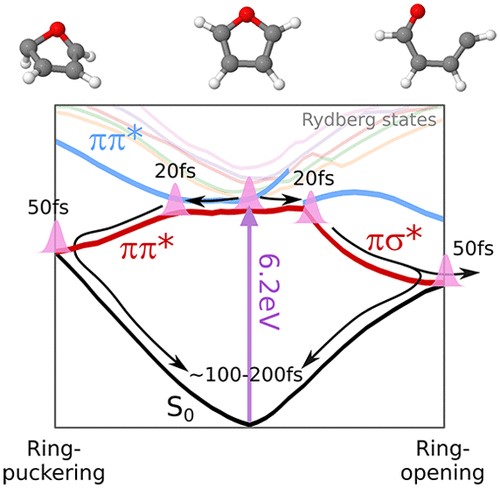
Ultrafast internal conversion of furan upon deep UV excitation at 200 nm is studied by using extreme ultraviolet time-resolved photoelectron spectroscopy with a time resolution of 15 fs. Ballistic nuclear wavepacket motion from the 1B2(π,π*) state to the ground state is fully observed using 21.7 eV probe pulses. Through the performance of a comparison with the results of electronic structure calculations at the MS(3)-CASPT2(10,10)/cc-pVTZ level of theory, the photoelectron signals from the conical intersection regions are identified.
Ultrafast Ring Closure Reaction of Gaseous cis-Stilbene from S1(ππ*)
Shutaro Karashima, Xincheng Miao, Akio Kanayama, Yo-ichi Yamamoto, Junichi Nishitani, Nikita Kavka, Roland Mitric, and Toshinori Suzuki
J. Am. Chem. Soc., 145, 6, 3283-3288 (2023)
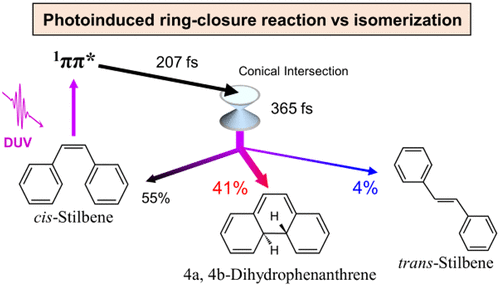
cis-Stilbene (cis-St) is a well-known benchmark system for cis–trans photoisomerization. cis-St also produces 4a,4b-dihydrophenanthrene (DHP) in solution with a quantum yield of less than 0.19. The ring closure reaction, however, has never been identified for gaseous cis-St, and a recent computational simulation predicted the quantum yield of DHP to be only 0.04. In the present study, we identified an ultrafast ring closure reaction of gaseous cis-St for the first time using extreme ultraviolet time-resolved photoelectron spectroscopy. Surface hopping trajectory calculations at the SA3-XMS-CASPT2(2,2) level of theory reproduce the features of the observed time-resolved photoelectron spectra and predict the cis-St:DHP:trans-St branching ratio to be 0.55:0.41:0.04, in contrast with previous estimates. The results indicate that photoexcited cis-St favors ring closure over cis–trans isomerization under the isolated condition.
Ultrafast ring-opening reaction of 1,3-cyclohexadiene: identification of non-adiabatic pathway via doubly excited state
Shutaro Karashima, Alexander Humeniuk, Ryuta Uenishi, Takuya Horio, Manabu Kanno, Tetsuro Ohta, Junichi Nishitani, Roland Mitrić, and Toshinori Suzuki
J. Am. Chem. Soc., 143, 21, 8034-8045 (2021)
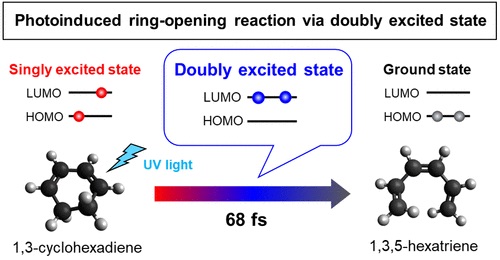
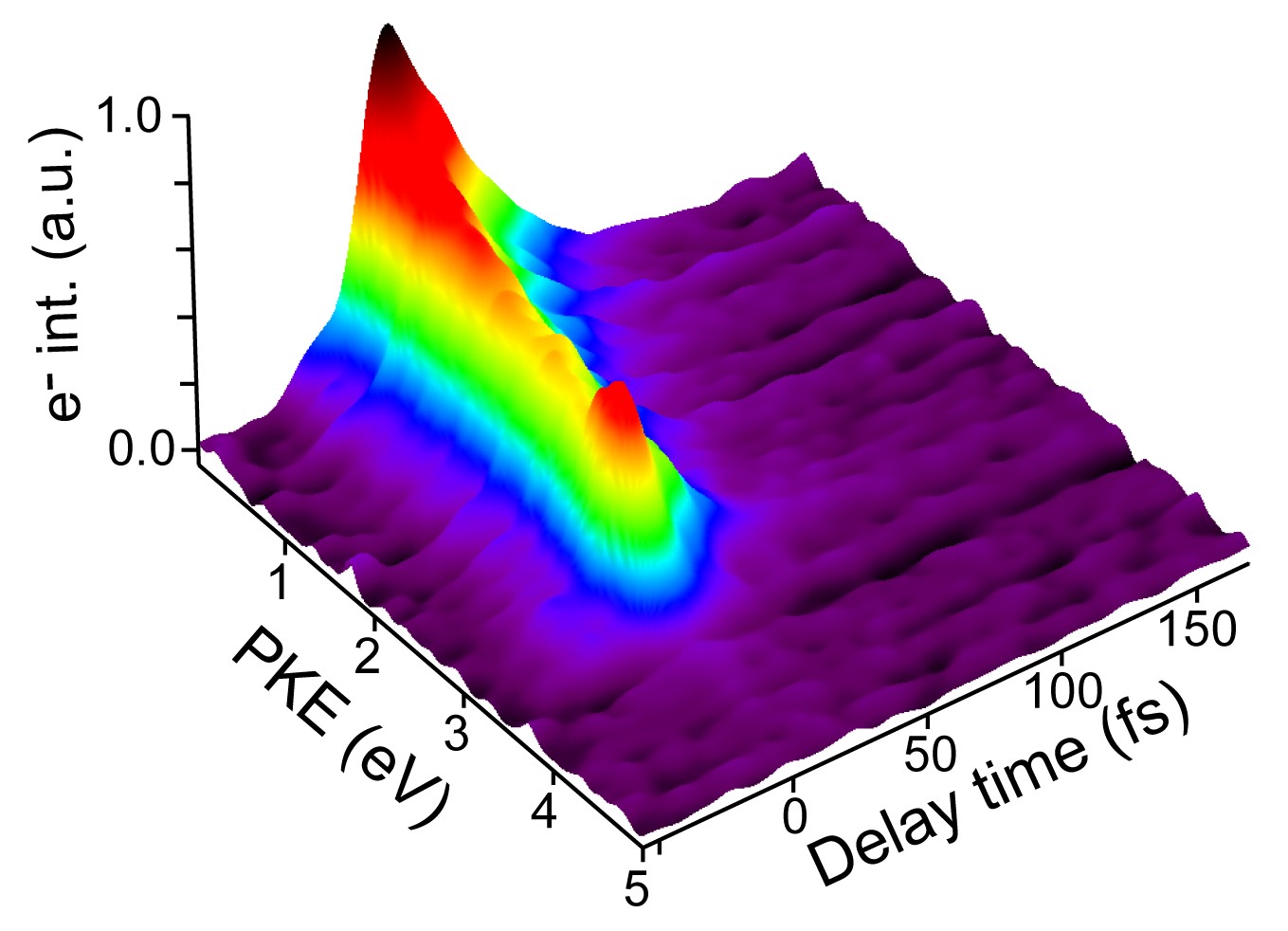
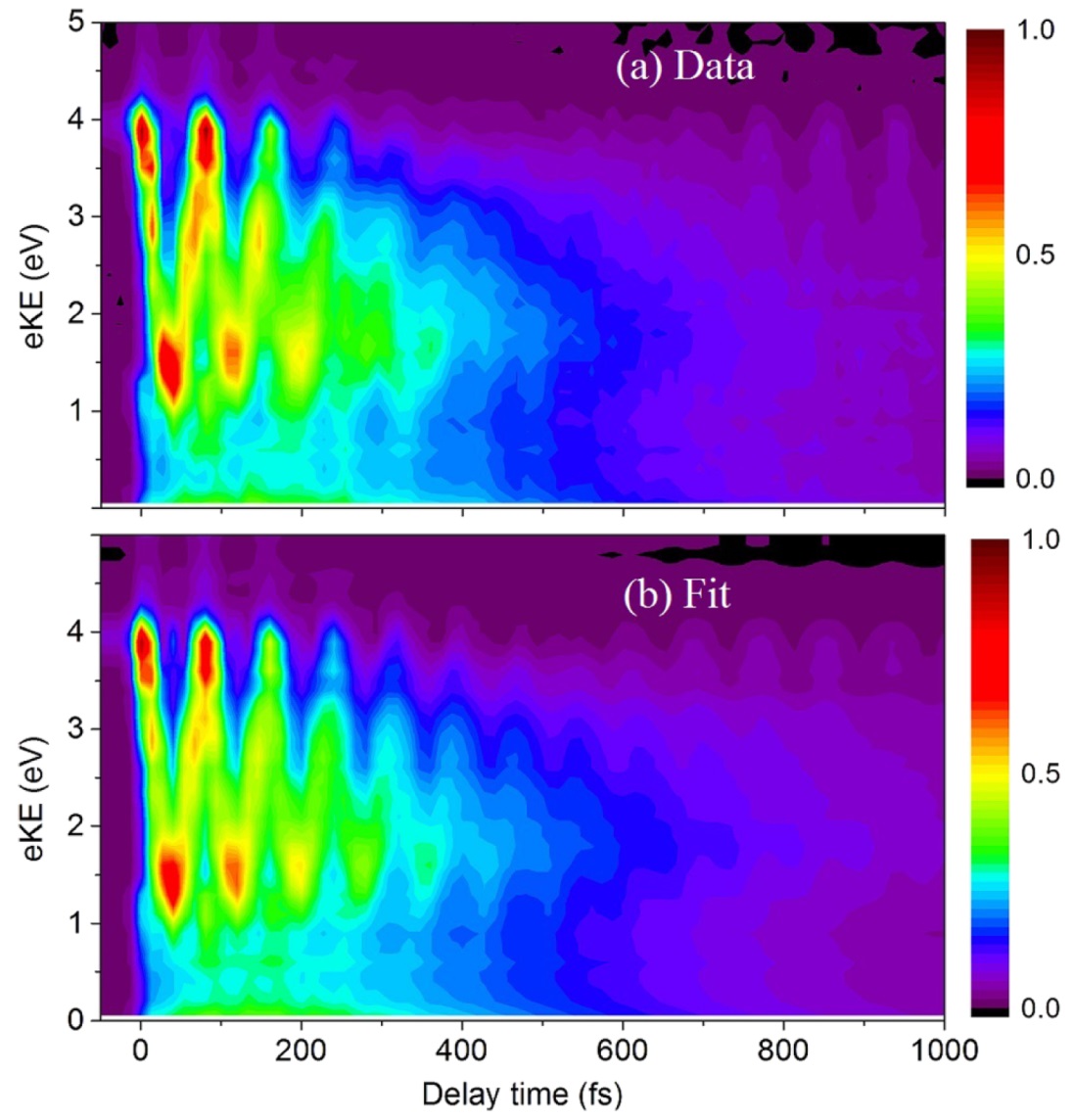
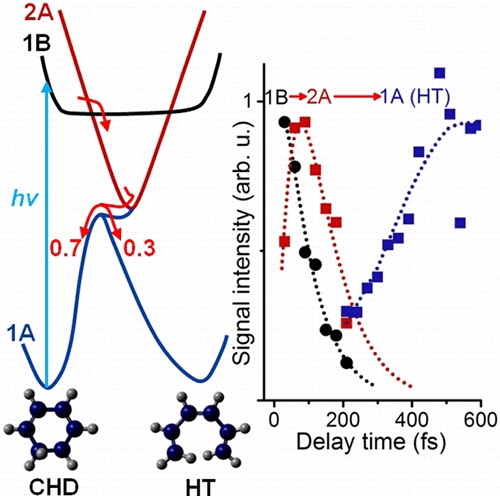
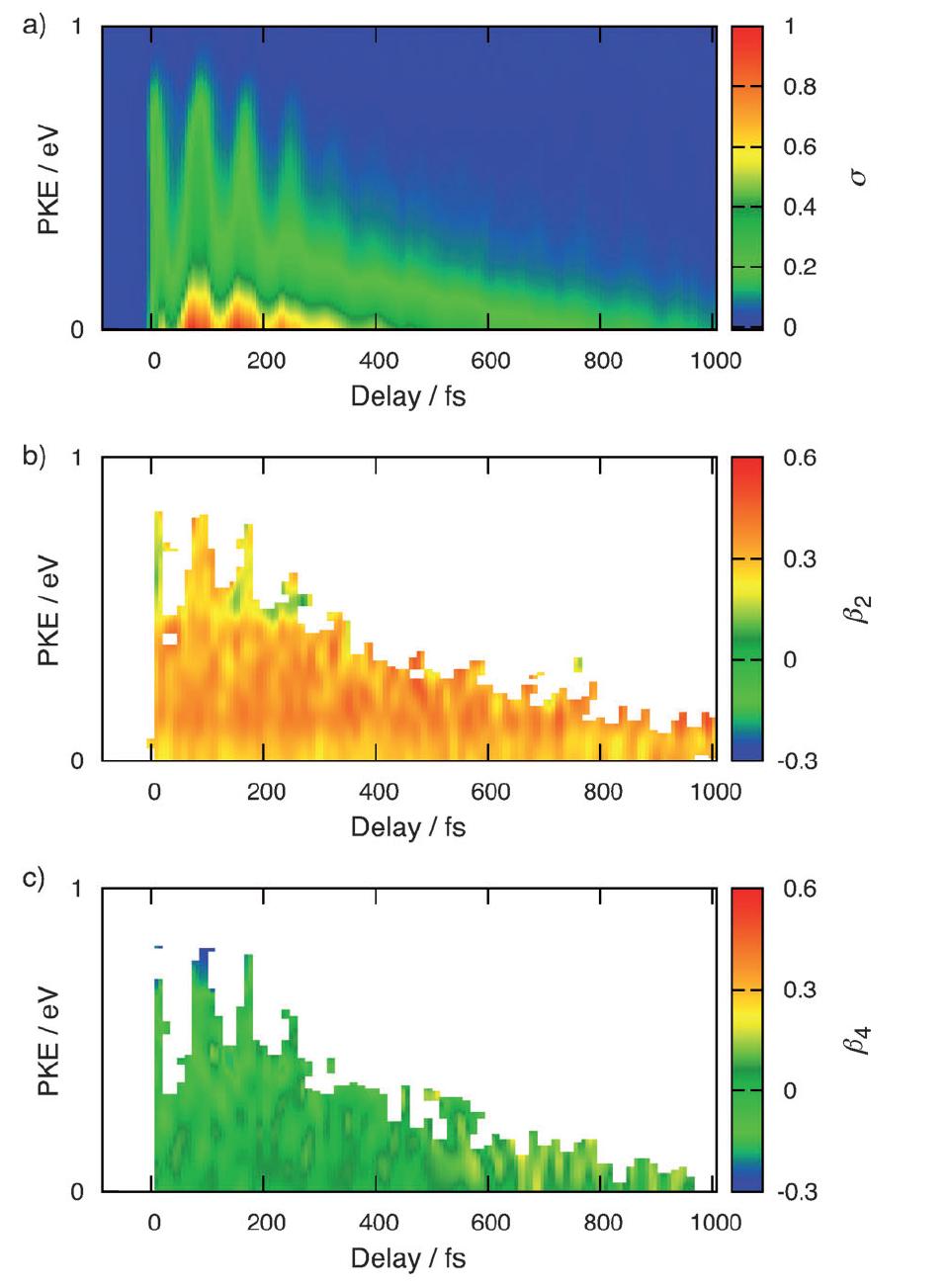
The photoinduced ring-opening reaction of 1,3-cyclohexadiene (CHD) to produce 1,3,5-hexatriene (HT) plays an essential role in the photobiological synthesis of vitamin D3 in the skin. This reaction follows the Woodward−Hoffmann rule, and C5−C6 bond rupture via an electronically excited state occurs with conrotatory motion of the end CH2 groups. However, it is noted that the photoexcited S1(π,π*) state of CHD is not electronically correlated with the ground state of HT, and the reaction must proceed via nonadiabatic transitions. In the present study, we have clearly observed the nonadiabatic reaction pathway via the doubly excited state of CHD using ultrafast extreme UV photoelectron spectroscopy. The results indicate that the reaction occurs in only 68 fs and creates product vibrational coherence. Extensive computational simulations support the interpretation of experimental results and provide further insights into the electronic dynamics in this paradigmatic electrocyclic ring-opening reaction.
Ultrafast extreme ultraviolet photoelectron spectroscopy of non-adiabatic photodissociation of CS2 from 1B2 (1Σu+) state: product formation via an intermediate electronic state
Shutaro Karashima, Yoshi-ichi Suzuki, and Toshinori Suzuki
J. Phys. Chem. Lett., 12, 3755-3761 (2021)
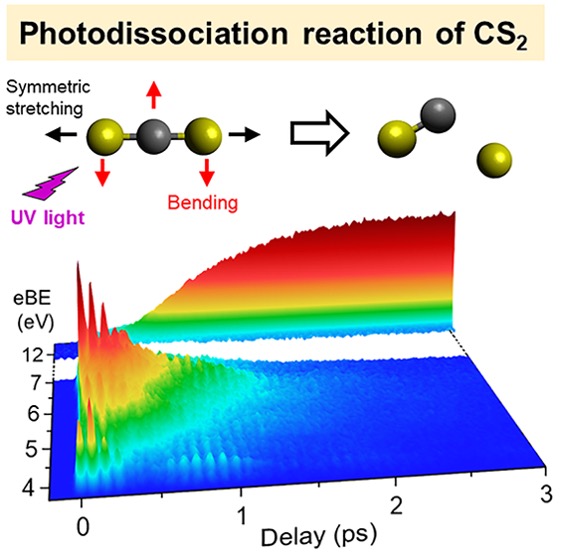
We studied nonadiabatic dissociation of CS2 from the 1B2 (1Σu+) state using ultrafast extreme ultraviolet photoelectron spectroscopy. A deep UV (200 nm) laser using the filamentation four-wave mixing method and an extreme UV (21.7 eV) laser using the high-order harmonic generation method were employed to achieve the pump−probe laser cross-correlation time of 48 fs. Spectra measured with a high signal-to-noise ratio revealed clear dynamical features of vibrational wave packet motion in the 1B2 state; its electronic decay to lower electronic state(s) within 630 fs; and dissociation into S(1D2), S(3PJ), and CS fragments within 300 fs. The results suggest that both singlet and triplet dissociation occur via intermediate electronic state(s) produced by electronic relaxation from the 1B2 (1Σu+) state.
Methyl substitution effects on non-adiabatic dynamics of benzene: lifting three-state quasi-degeneracy at conical intersections
Shunsuke Adachi and Toshinori Suzuki
Phys. Chem. Chem. Phys., 22, 2814-2818 (2020)
Previously, theoretical calculations on the non-adiabatic dynamics of benzene from the S2 state have indicated that the S2/S1 and S1/S0 conical intersections (CIs) facilitate ballistic nuclear wavepacket motion from S2 to S0 (fast channel) and branching to S1 (slow channel). In this paper, we present time-resolved photoelectron spectra of benzene and its methyl-derivatives (toluene and o-xylene) measured with a vacuum-UV laser, which clearly reveal both the fast and slow channels. The extremely short propagation time of the wavepacket between the two CIs of benzene indicates that the two are in close proximity to each other, while methyl substitution extends the propagation time and decreases the branching ratio into the fast channel. The results suggest that the quasi-degeneracy of the three states in benzene is lifted by the geometrical shifts of the CIs by methyl substitution.
Time-resolved photoelectron imaging of acetone with 9.3 eV photoexcitation
Ryuta Uenishi, Takuya Horio, and Toshinori Suzuki
J. Phys. Chem. A, 123, 6848-6853 (2019)
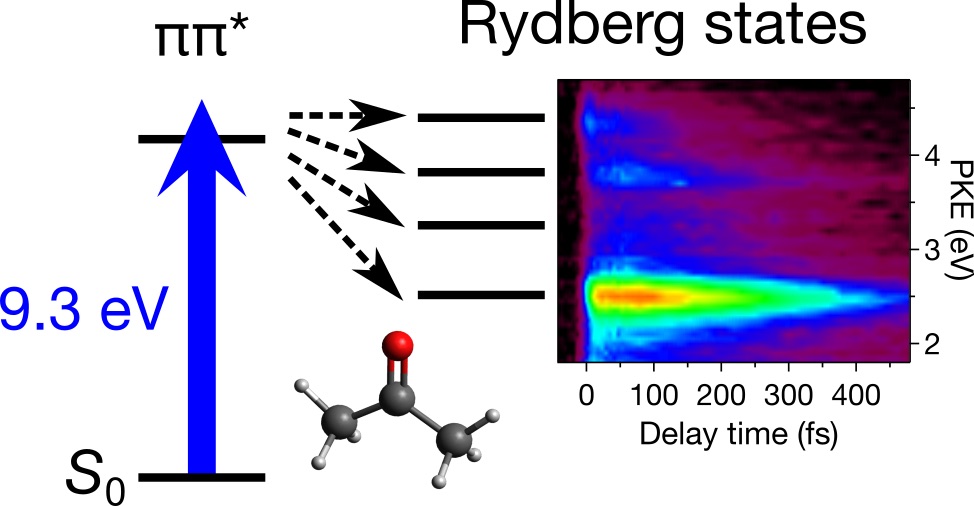
Ultrafast electronic relaxation following 9.3 eV photoexcitation of gaseous acetone was investigated with time-resolved photoelectron imaging spectroscopy. An intense photoionization signal due to a transition from the 41A1(π,π*) state to the D1(π−1) cationic state diminishes within 50 fs, owing to vibrational wave packet motion leaving our observation energy window. Additional photoionization signals were assigned to transitions from Rydberg states with principal quantum numbers of 3−8 to the D0(n−1) cationic state, created by strong vibronic couplings with the bright 41A1(π,π*) state. The deactivation processes of the 41A1(π,π*) and Rydberg states are discussed based on their decay profiles obtained from a time−energy map of photoelectron kinetic energy distributions.
Probing ultrafast dynamics during and after passing through conical intersections
Shunsuke Adachi, Tom Schatteburg, Alexander Humeniuk, Roland Mitrić, and Toshinori Suzuki
Phys. Chem. Chem. Phys. , 21, 13902-13905 (2019)
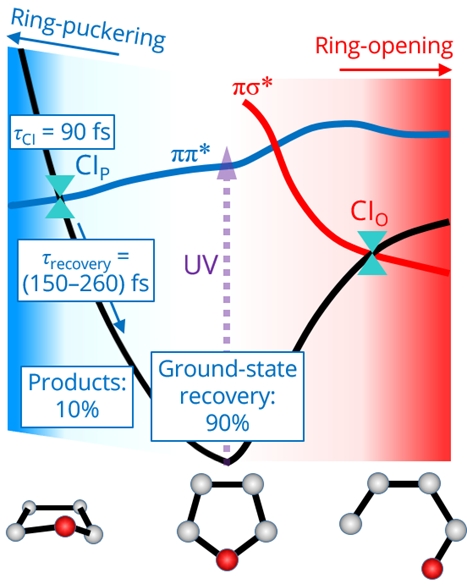
Time-resolved photoelectron spectroscopy using vacuum-UV probe pulses enables observing ultrafast dynamics during and after passing through conical intersections (CIs). The ring-puckering CI plays a prominent role following the ππ* photoexcitation of furan. More than 90% of the excited molecules safely return to the original ground state, while the remaining 10% transforms into isomers after passing through the puckering CI.
Unravelling the electronic state of NO2 product in ultrafast photodissociation of nitromethane
Shunsuke Adachi, Hiroshi Kohguchi, and Toshinori Suzuki
J. Phys. Chem. Lett., 9, 270-273 (2018)
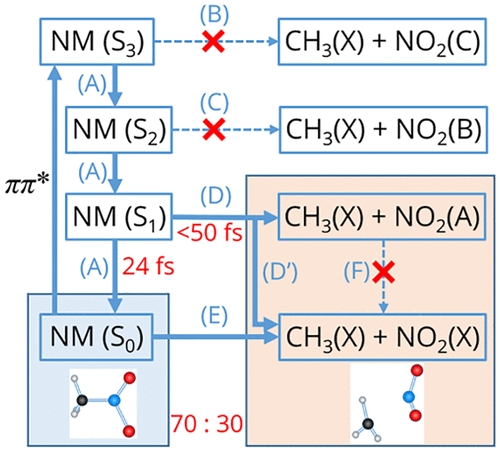
The primary photochemical rezction of nitromethane (NM) after ππ* exicitation is known to be C-N bond cleavage (CH3NO2 + hν → CH3 + NO2). On the other hand, NO2 can be formed in both the ground and exited states, and identification of the electronic state of the NO2 product has been a central subject in the experimental and theoretical studies. Here we present time-resolved photoelectron spectroscopy using vacuum-ultraviolet probe pulses to observe all transient electronic states of NM and the reaction products. The result indicates that ultrafast internal conversion occurs down to S1 and S0 within 24 fs, and the dissociation proceeds on the S1 surface (τdiss ≲ 50 fs), leading to comparable product yields of NO2(A) and NO2(X). The overall dissociation quantum yield within our observation time window (< 2 ps) is estimated to be 0.29.
Real-time detection of S(1D2) photofragments produced from the 1B2(1Σu+) state of CS2 by vacuum ultraviolet photoelectron imaging using 133 nm probe pulses
Takuya Horio, Roman Spesyvtsev, Yu Furumido, and Toshinori Suzuki
J. Chem. Phys., 147, 013932 (2017)
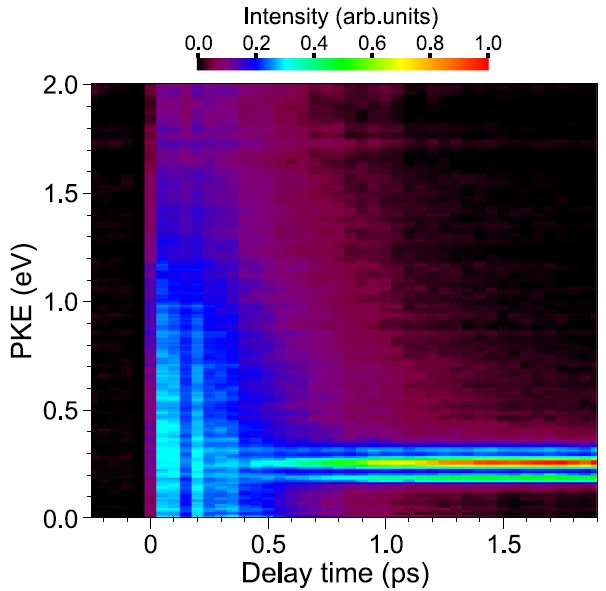
Ultrafast photodissociation dynamics from the 1B2(1Σu+) state of CS2 are studied by time-resolved photoelectron imaging using the fourth (4ω, 198 nm) and sixth (6ω, 133 nm) harmonics of a femtosecond Ti:sapphire laser. The 1B2 state of CS2 was prepared with the 4ω pulses, and subsequent dynamics were probed using the 6ω vacuum ultraviolet (VUV) pulses. The VUV pulses enabled real-time detection of S(1D2) photofragments, produced via CS2*(1B2(1Σu+)) → CS(X 1Σ+) + S(1D2). The photoionization signal of dissociating CS2*(1B2(1Σu+)) molecules starts to decrease at about 100 fs, while the S(1D2) fragments appear with a finite (ca. 400 fs) delay time after the pump pulse. Also discussed is the configuration interaction of the 1B2(1Σu+) state based on relative photoionization cross-sections to different cationic states.
Unexpectedly broad photoelectron spectrum as a signature of ultrafast electronic relaxation of Rydberg states of carbon dioxide
Shunsuke Adachi, Motoki Sato, Toshinori Suzuki, and Sergy Yu. Grebenshchikov
Phys. Rev. A, 95, 033422 (2017)
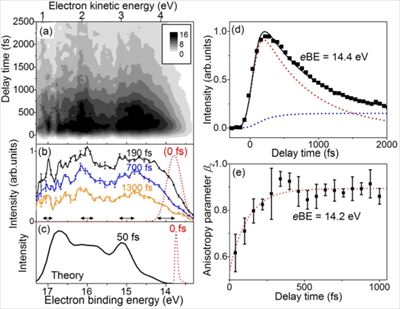
The dynamics of CO2 excited into Rydberg states lying 0.2 eV below the ionization threshold is studied by means of time resolved photoelectron imaging. Over 3 eV broad photoelectron spectra are measured for all pump-probe delay times. Quantum mechanical calculations demonstrate that the spectral broadening is due to ultrafast electronic relaxation of Rydberg states and identify the likely relaxation pathways. Experiment and theory bracket the relaxation time between 15 and 65 fs. A weak time independent ionization signal is attributed to CO2 trapped in near-threshold triplet states.
Ultrafast photodynamics of pyrazine in the vacuum ultraviolet region studied by time-resolved photoelectron imaging using 7.8-eV pulses
Takuya Horio, Yoshi-ichi Suzuki, and Toshinori Suzuki
J. Chem. Phys., 145, 044307 (2016)
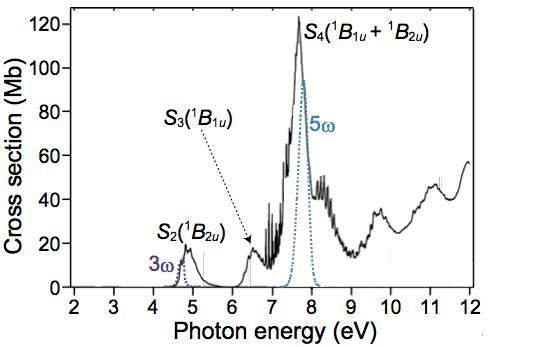
The ultrafast electronic dynamics of pyrazine (C4N2H4) were studied by time-resolved photoelectron imaging (TRPEI) using the third (3ω, 4.7 eV) and fifth harmonics (5ω,7.8 eV) of a femtosecond Ti:sapphire laser (ω). Although the photoionization signals due to the 5ω - 3ω and 3ω - 5ω pulse sequences overlapped near the time origin, we have successfully extracted their individual TRPEI signals using least squares fitting of the observed electron kinetic energy distributions. When the 5ω pulses preceded the 3ω pulses, the 5ω pulses predominantly excited the S4 (ππ*,1B1u + 1B2u) state. Photoionization signal from the S4 state generated by the time-delayed 3ω pulses was dominated by the D3(2B2g) ← S4 photoionization process and exhibited a broad electron kinetic energy distribution, which rapidly downshifted in energy within 100 fs. Also observed were photoionization signals for the 3s, 3pz, and 3py members of the Rydberg series converging to D0(2Ag). The Rydberg signals appeared immediately within our instrumental time resolution of 27 fs, indicating that these states are directly photoexcited from the ground state or populated from S4 within 27 fs. The 3s, 3pz, and 3py states exhibited single exponential decay with lifetimes of 94 ± 2, 89 ± 2, and 58 ± 1 fs, respectively. With the reverse pulse sequence of 3ω - 5ω, the ultrafast internal conversion (IC) from S2(ππ*) to S1(nπ*) was observed. The decay associated spectrum of S2 exhibited multiple photoionizations to D0, D1, and D3, in agreement with the 3ω-pump and 6ω-probe experiment described in our preceding paper. The electron kinetic energy and angular distributions from S1 populated by IC from S2 are also discussed.
Full observation of ultrafast cascaded radiationless transitions from S2(ππ*) state of pyrazine using vacuum ultraviolet photoelectron imaging
Takuya Horio, Roman Spesyvtsev, Kazuki Nagashima, Rebecca A. Ingle, Yoshi-ichi Suzuki, and Toshinori Suzuki
J. Chem. Phys., 145, 044306 (2016)
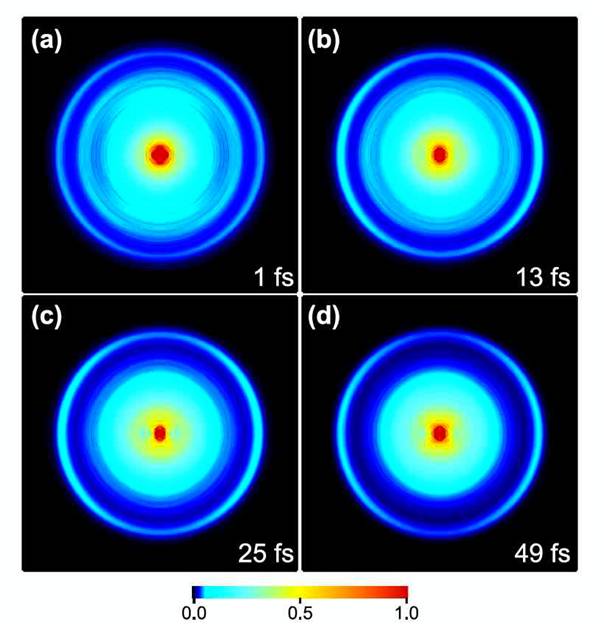
A photoexcited molecule undergoes multiple deactivation and reaction processes simultaneously or sequentially, which have been observed by combinations of various experimental methods. However, a single experimental method that enables complete observation of the photo-induced dynamics would be of great assistance for such studies. Here we report a full observation of cascaded electronic dephasing from S2(ππ*) in pyrazine (C4N2H4) by time-resolved photoelectron imaging (TRPEI) using 9.3-eV vacuum ultraviolet (VUV) pulses with a sub-20 fs time duration. While we previously demonstrated a real-time observation of the ultrafast S2(ππ*)→S1(nπ*) internal conversion in pyrazine using TRPEI with UV pulses, this study presents complete observation of the dynamics including radiationless transitions from S1 to S0 (internal conversion) and T1(nπ*) (intersystem crossing). Also discussed are the role of 1Au(nπ*) in the internal conversion and the configuration interaction of the S2(ππ*) electronic wave function.
Photoisomerization of vibrationally hot tetramethylethylene produced by ultrafast internal conversion from the excited state
Motoki Sato, Shunsuke Adachi, and Toshinori Suzuki
J. Phys. Chem. A, 120, 5099 - 5102 (2016)
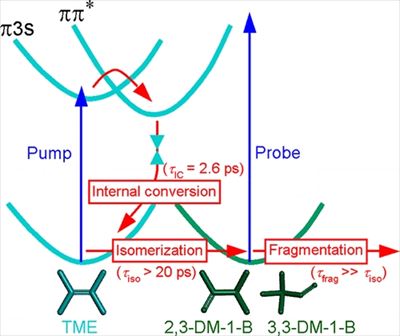
Isomerization of tetramethylethylene (TME) following ultrafast internal conversion was investigated using time-resolved photoelectron spectroscopy with vacuum-ultraviolet probe pulses. The difference photoelectron spectrum at τ = 15 ps was reasonably well reproduced using a linear combination of static photoelectron spectra of TME and its isomers. The isomers were produced as a consequence of unimolecular reaction of vibrationally hot TME, created by internal conversion from the excited state.
Linear and Circular Dichroism in Photoelectron Angular Distributions Caused by Electron Correlation
Yoshi-Ichi Suzuki and Toshinori Suzuki
Phys. Rev. A, 91, 053413 (2015)
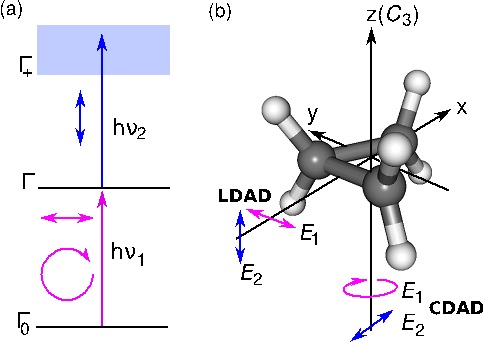
Electron correlation can break the symmetry of photoelectron angular distribution upon two step ionization to a doubly degenerate ionic state via a doubly degenerate intermediate state. The interference between the two components of the intermediate state prepared using linearly and circularly polarized light is discussed for cyclopropane as an example.
Ultrafast deactivation of the ππ*(V) state of ethylene studied using sub-20 fs time-resolved photoelectron imaging
Takufumi Kobayashi, Takuya Horio, and Toshinori Suzuki
J. Phys. Chem. A, 119 (36), 9518-9523 (2015)
The ultrafast deactivation process of ethylene in the ππ*(V) state was studied using time-resolved photoelectron imaging with sub-20 fs pulses at 159 and 198 nm. The photoelectron kinetic energy distribution observed upon 159 nm photoexcitation exhibited a continuous downward shift within 20 fs, attributed to both C-C twist and pyramidalization motions. A partial revival of the vibrational wave packet was observed with the period of about 18 fs, which is attributed to the C-C twist from 0 to 180° on the ππ*(V) potential energy surface. Signature for internal conversion from the ππ*(V) state to a lower-lying π3s Rydberg (R) state, which has been previously suggested, was not detected in the time-dependent photoelectron kinetic energy and angular distributions.
Observation of the wavepacket dynamics on the 1B2(1Σu+) state of CS2 by sub-20 fs photoelectron imaging using 159 nm probe pulses
Roman Spesyvtsev, Takuya Horio, Yoshi-ichi Suzuki, and Toshinori Suzuki
J. Chem. Phys., 142, 074308 (2015)
The wavepacket dynamics of CS2 after photoexcitation to the 1B2(1Σu+) state at 198 nm are studied by time-resolved photoelectron imaging using sub-20 fs 159 nm pulses, which enable single photon ionization from the entire region of the 1B2 potential energy surface. The time-energy map of the photoelectron intensity reveals vibrational motions along the symmetric stretching and bending coordinates. The time-energy map of the photoelectron anisotropy parameter exhibits time-evolution within single oscillation periods of the ν1 and ν2 modes, which is attributed to variation of the excited state electronic character along these vibrational coordinates. The initially populated 1B2 state evolves with two time constants of 107 and 394 fs.
Direct Observation of Ground-State Product Formation in a 1,3-Cyclohexadiene Ring-Opening Reaction
Shunsuke Adachi, Motoki Sato, and Toshinori Suzuki
J. Phys. Chem. Lett., 6 (3), 343-346 (2015)
Ultrafast photoelectron imaging using a 90 nm vacuum-UV probe pulse is applied to the ring-opening reaction of 1,3-cyclohexadiene (CHD) in the gas phase, and formation of 1,3,5-hexatriene (HT) and CHD in their electronic ground states is observed in real time. The analysis of the transient photoelectron kinetic energy spectra reveals the branching ratio into HT and CHD as 3:7 upon 270 nm photoexcitation. The ratio is in reasonable agreement with the experimental values reported for the liquid phase and theoretical values for the gas phase, resolving the discrepancy.
Excited-State Dynamics of CS2 Studied by Photoelectron Imaging with a Time Resolution of 22 fs
Takao Fuji, Yoshi-Ichi Suzuki, Takuya Horio, and Toshinori Suzuki
Chem. Asian J., 6, 3028-3034 (2011)
The ultrafast dynamics of CS2 in the 1B2(1Σu+) state was studied by photoelectron imaging with a time resolution of 22 fs. The photoelectron signal intensity exhibited clear vibrational quantum beats due to wave packet motion. The signal intensity decayed with a lifetime of about 400 fs. This decay was preceded by a lag of around 30 fs, which was considered to correspond to the time for a vibrational wave packet to propagate from the Franck-Condon region to the region where predissociation occurred. The photoelectron angular distribution remained constant when the pump-probe delay time was varied. Consequently, variation of the electronic character caused by the vibrational wave packet motion was not identified within the accuracy of our measurements.
Time-resolved photoelectron imaging of S2 → S1 internal conversion in benzene and toluene
Yoshi-ichi Suzuki, Takuya Horio, Takao Fuji, and Toshinori Suzuki
J. Chem. Phys., 134, 184313 (2011)
Ultrafast internal conversion of benzene and toluene from the S2 states was studied by time-resolved photoelectron imaging with a time resolution of 22 fs. Time-energy maps of the photoelectron intensity and the angular anisotropy were generated from a series of photoelectron images. The photoelectron kinetic energy distribution exhibits a rapid energy shift and intensity revival, which indicates nuclear motion on the S2 adiabatic surface, while the ultrafast evolution of the angular anisotropy revealed a change in the electronic character of the S2 adiabatic surface. From their decay profiles of the total photoelectron intensity, the time constants of 48 ± 4 and 62 ± 4 fs were determined for the population decay from the S2 states in benzene and toluene, respectively.
He(I) Ultraviolet Photoelectron Spectroscopy of Benzene and Pyridine in Supersonic Molecular Beams Using Photoelectron Imaging
Suet Yi Liu, Koutayba Alnama, Jun Matsumoto, Kiyoshi Nishizawa, Hiroshi Kohguchi, Yuan-Pern Lee, and Toshinori Suzuki
J. Phys. Chem. A, 115 (14), 2953-2965 (2011)
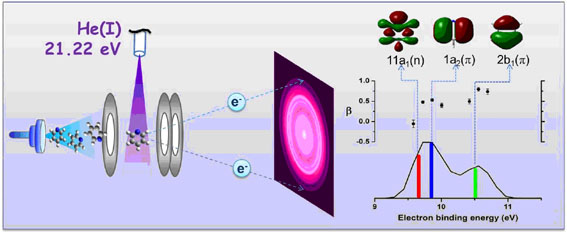
We performed He I ultraviolet photoelectron spectroscopy (UPS) of jet-cooled aromatic molecules using a newly developed photoelectron imaging (PEI) spectrometer. The PEI spectrometer can measure photoelectron spectra and photoelectron angular distributions at a considerably higher efficiency than a conventional spectrometer that uses a hemispherical energy analyzer. One technical problem with PEI is its relatively high susceptibility to background electrons generated by scattered He I radiation. To reduce this problem, we designed a new electrostatic lens that intercepts background photoelectrons emitted from the repeller plate toward the imaging detector. An energy resolution (ΔE/E) of 0.735% at E = 5.461 eV is demonstrated with He I radiation. The energy resolution is limited by the size of the ionization region. Trajectory calculations indicate that the system is capable of achieving an energy resolution of 0.04% with a laser if the imaging resolution is not limited. Experimental results are presented for jet-cooled benzene and pyridine, and they are compared with results in the literature.
Photoelectron Imaging Spectroscopy of S1(1B2u ππ*) Benzene via 611n (n=0-3) Levels
Dongmei Niu, Yoshihiro Ogi, Yoshi-Ichi Suzuki, and Toshinori Suzuki
J. Phys. Chem. A, 115 (11), 2096-2102 (2011)
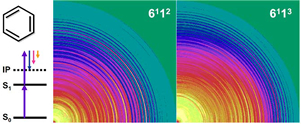
We report resonance-enhanced two-photon ionization photoelectron spectroscopy of jet-cooled Benzene via the 611n (n = 0-3) vibronic levels in S1(1B2u ππ*) using a nanosecond UV laser and photoelectron imaging. The best energy resolution (ΔE/E) was 0.7 %. The photoelectron spectrum from the S1 6113 level (Evib = 3284 cm-1) in the channel three region exhibited a clear signature of intramolecular vibrational redistribution (IVR). The spectral features were consistent with picosecond zero kinetic energy photoelectron (ZEKE) spectra reported by Smith et al. [J. Phys. Chem. 1995, 99, 1768]. The photoelectron angular anisotropy parameter b2 was found to be negative in ionization from the 611n (n = 0-3) levels with photoelectron kinetic energies up to 5000 cm-1. No influence of a shape resonance was identified.
Ultrafast photodynamics of furan
Takao Fuji, Yoshi-Ichi Suzuki, Takuya Horio, Toshinori Suzuki, Roland Mitrić, Ute Werner, and Vlasta Bonačić-Koutecký
J. Chem. Phys., 133, 234303 (2010)
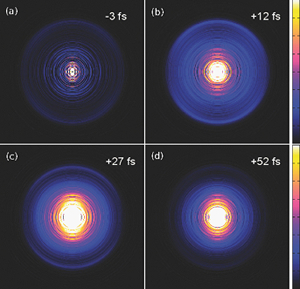
Ultrafast photodynamics of furan has been studied by time-resolved photoelectron imaging (TRPEI) spectroscopy with an unprecedented time resolution of 22 fs. The simulation of the time-dependent photoelectron kinetic energy distribution (PKED) has been performed with ab initio nonadiabatic dynamics “on the fly” in the frame of time-dependent density functional theory. Based on the agreement between experimental and theoretical time-dependent photoelectron signal intensity as well as on PKED, precise time scales of ultrafast internal conversion from S2 over S1 to the ground state S0 of furan have been revealed for the first time. Upon initial excitation of the S2 state which has π-π* character, a nonadiabatic transition to the S1 state occurs within 10 fs. Subsequent dynamics invokes the excitation of the C-O stretching and C-O-C out of plane vibrations which lead to the internal conversion to the ground state after 60 fs. Thus, we demonstrate that the TRPEI combined with high level nonadiabatic dynamics calculations provide fundamental insight into ultrafast photodynamics of chemically and biologically relevant chromophores.
Time-resolved photoelectron imaging of ultrafast S2→S1 internal conversion through conical intersection in pyrazine
Yoshi-Ichi Suzuki, Takao Fuji, Takuya Horio, and Toshinori Suzuki
J. Chem. Phys., 132, 174302 (2010)
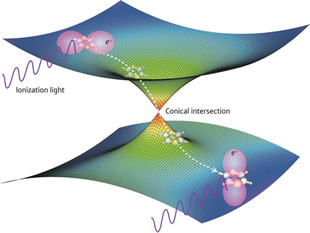
A nonadiabatic electronic transition through a conical intersection was studied by pump-probe photoelectron imaging spectroscopy with a 22 fs time resolution in the benchmark polyatomic molecule of pyrazine and deuterated pyrazine. The lifetimes of the S2 state of pyrazine and deuterated pyrazine were determined to be 22±3 fs by the global fitting of the time-energy maps of photoelectron kinetic energy (PKE) distributions. The lifetime of S3 was determined to be 40-43 fs. Two-dimensional maps of photoelectron distributions were obtained for time (t) and PKE, and individual PKE distributions upon ionization from S2 and S1 were extracted. Quantum beat with an approximately 50 fs period was observed after the S2→S1 internal conversion, which was attributed to the totally symmetric vibration v 6a in S1.
Time-resolved photoelectron imaging using a femtosecond UV laser and a VUV free-electron laser
S. Y. Liu, Y. Ogi, T. Fuji, K. Nishizawa, T. Horio, T. Mizuno, H. Kohguchi, M. Nagasono, T. Togashi, K. Tono, M. Yabashi, Y. Senba, H. Ohashi, H. Kimura, T. Ishikawa, and T. Suzuki
Phys. Rev., 81, 031403 (2010)
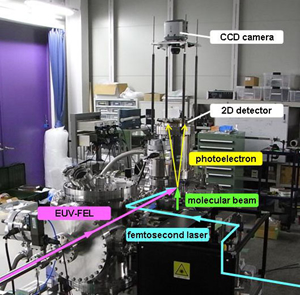
A time-resolved photoelectron imaging using a femtosecond ultraviolet (UV) laser and a vacuum UV freeelectron laser is presented. Ultrafast internal conversion and intersystem crossing in pyrazine in a supersonic molecular beam were clearly observed in the time profiles of photoioinzation intensity and time-dependent photoelectron images.
Molecular frame image restoration and partial wave analysis of photoionization dynamics of NO by time-energy mapping of photoelectron angular distribution
Ying Tang, Yoshi-Ichi Suzuki, Takuya Horio, and Toshinori Suzuki
Phys. Rev. Lett., 104, 073002 (2010)
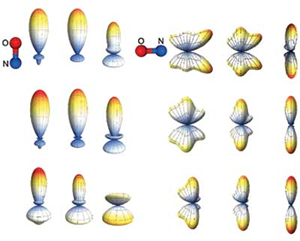
The benchmark system of molecular photoionization dynamics, the (1 + 1') two-photon ionization of NO via the A state, is investigated using the time-energy mapping of the photoelectron angular distribution in a laboratory frame. The molecular frame photoelectron angular distribution and partial wave composition are determined from time-energy maps and compared with those obtained by Schwinger variational calculation (SVC) and state-to-state photoelectron spectroscopy. Good agreement is found with SVC. By comparison of the phase shifts of the scattering waves and the quantum defects of the Rydberg states, the l hybridization of p waves is identified.
Super-Resolution Photoelectron Imaging with Real-Time Subpixelation by Field Programmable Gate Array and Its Application to NO and Benzene Photoionization
Yoshihiro Ogi, Hiroshi Kohguchi, Dongmei Niu, Keijiro Ohshimo, and Toshinori Suzuki
J. Phys. Chem. A, 113 (52), 14536-14544 (2009)

We have constructed a photoelectron imaging spectrometer with super-resolution image processing and have applied it to the photoionization of nitric oxide and benzene in molecular beams. A field programmable gate array is employed for real-time subpixel centroiding calculations on hardware, providing 64 megapixel resolution (8192 × 8192 pixels). We examined eight different centroiding algorithms based on the center-of-gravity (COG) and Gaussian fitting (Gauss) methods and have found that the two-dimensional COG (2D-COG) and weighted mean of Gaussian center (w-Gauss) methods have the best performance. The excellent performance of the instrument is demonstrated by visualizing a 25 μm diameter pore structure of an MCP, indicating a spatial resolution of 0.03%. The photoelectron image in one-color (1 + 1) resonance-enhanced multiphoton ionization of nitric oxide using a nanosecond laser provided a photoelectron kinetic energy resolution of 0.2%. This resolution is currently restricted by charged-particle optics. The photoelectron energy and angular distributions in the one-color (1 + 1) resonance-enhanced multiphoton ionization of benzene via 61 and 6111 vibronic levels in the S1 state are also presented. The results demonstrate that photoelectron angular anisotropy varies with the photoelectron kinetic energy and the vibronic state of the cation.
3. Transient Absorption Spectroscopy
A Twist in Electronic Relaxation of Pyrimidine Nucleosides and Nucleotides: Impact of C5 Methylation on Nonadiabatic Transition and Photohydration Damage
Srijon Ghosh, Yuki Obara, Vishal Kumar Jaiswal, Mario Taddei, Artur Nenov, Irene Conti, Marco Garavelli and Toshinori Suzuki
J. Am. Chem. Soc., 148, 18903-18918 (2026)
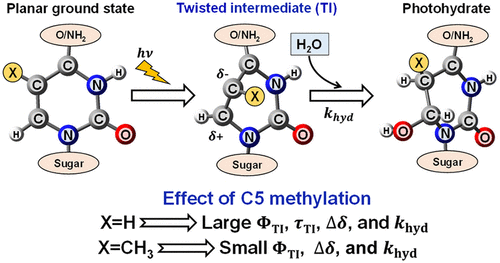
We report a combined experimental and theoretical study of the formation and decay dynamics of a ground-state twisted intermediate (TI) involved in the ultrafast internal conversion of UV-excited pyrimidine nucleosides and nucleotides in aqueous solution. Infrared transient absorption spectroscopy identifies a TI featuring a strongly twisted C5=C6 double bond, and its quantum yield ΦTI and lifetime τTI are determined for structurally distinct nucleosides and nucleotides. Photohydration rates khyd measured under continuous 266 nm irradiation correlate directly with ΦTI × τTI, providing unambiguous evidence that the TI mediates the hydration reaction. Comparison of C5–H and C5–CH3 derivatives reveals pronounced reductions in both ΦTI and khyd upon methylation. Quantum mechanics/molecular mechanics dynamical simulations show that TI formation requires sufficient nuclear momentum along the TI-forming coordinate at the conical intersection, whereas vibrational energy randomization induced by C5 methylation and solvent interactions diminishes this momentum and consequently ΦTI. The TI is neither diradical nor zwitterionic but instead contains an elongated, highly reactive C5=C6 double bond whose polarization and hydration reactivity are attenuated by C5 methylation. Consistently, the normalized reactivity index, khyd/(ΦTI × τTI), is substantially lower for C5-methylated compounds. A high activation barrier limits the photohydration quantum yield (∼0.01), and τTI is primarily governed by isomerization back to the planar ground-state structure.
Influence of C5 Methylation on Cytidine Photohydration Revealed by Deep-Ultraviolet Transient Absorption Anisotropy Spectroscopy
Shunsuke Adachi, Yuki Obara, Rin Shiomoto, and Toshinori Suzuki
J. Phys. Chem. Lett., 16, 11717-11723 (2025)
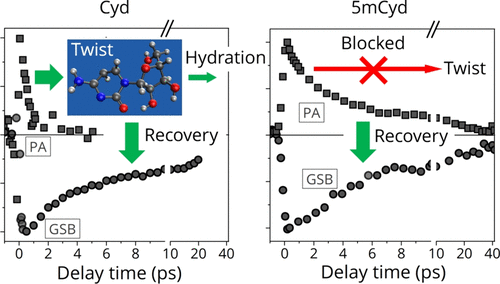
C5 methylation enhances the photostability of monomeric cytidine (Cyd), but the molecular basis of this effect has remained unresolved. Here, we examine the photodynamics of Cyd and its C5-methylated analogue, 5-methylcytidine (5mCyd), using femtosecond deep-ultraviolet transient absorption anisotropy spectroscopy. Recent theoretical and spectroscopic studies have highlighted a metastable ground-state intermediate with a twisted C5=C6 double bond, which plays a key role in the photodynamics of pyrimidine nucleobases. In Cyd, our anisotropy-resolved measurements captured the sub-picosecond 1ππ* → S0 internal conversion, with no detectable signature of a long-lived 1nπ* state. Instead, our results support the generation of a ground-state twisted conformer as the reactive intermediate for hydration. In 5mCyd, the yield of this intermediate is significantly reduced, indicating that C5 methylation suppresses its formation. The observed correlation between twisted conformer formation and the photostability toward hydration highlights how subtle chemical modifications can markedly alter photochemical activities.
Solvent Effects on the Formation of Ground-State Twisted Intermediate during Electronic Relaxation of Pyrimidine Nucleobases
Yuki Obara, Srijon Ghosh, Shota Kamibashira, and Toshinori Suzuki
J. Am. Chem. Soc., 147, 41284-41296 (2015)
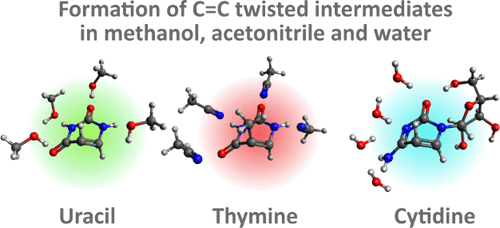
This study investigates solvent effects on the formation of ground-state twisted intermediates recently found in the electronic relaxation of uracil and thymine [Obara et al., J. Am. Chem. Soc. 2025, 147, 15077]. We newly identify a similar reaction intermediate characterized by a twisted C5=C6 bond for aqueous cytidine, establishing a structural motif common to all pyrimidine nucleobases and their derivatives. Unlike uracil and thymine, the cytidine intermediate lacks a blue-shifted band in its IR spectrum associated with C=O stretching, precluding its identification using the same spectral criteria applied to uracil and thymine. Nevertheless, quantum chemical calculations assuming a twisted geometry reproduce the experimental IR spectrum, notably identifying the 1580 cm-1 band previously misassigned to the 1nπ* state of cytidine as the marker band for the twisted intermediate. The marker band appears within 1 ps after UV excitation in polar protic solvents such as water and methanol, indicating that the twisted intermediate is generated directly from the 1ππ* state via ultrafast internal conversion through the ethylenic conical intersection. The polar aprotic solvent acetonitrile suppresses the formation of the twisted intermediate and increases the yield of the 3ππ* state for uracil and thymine. The primary difference between protic and aprotic solvents arises from the destabilization of the 1nπ* state in the former. The complexity of solvent effects warrants detailed theoretical modeling to elucidate the interplay between the solute and solvent in nonadiabatic dynamics in solution.
Formation of Ground-State Intermediate during Electronic Relaxation of Pyrimidine Nucleobases
Yuki Obara, Srijon Ghosh, Alexander Humeniuk, Shota Kamibashira, Shunsuke Adachi, and Toshinori Suzuki
J. Am. Chem. Soc., 147, 15077-15087 (2025)
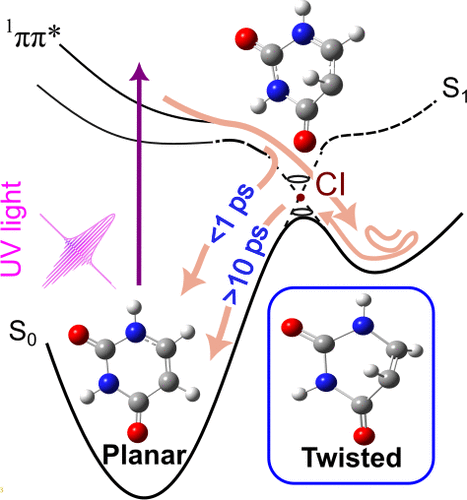
Ultrafast electronic relaxation of nucleobases from the 1ππ* state to the ground electronic state (S0) is crucial for the photostability of DNA and RNA. However, it has been suggested that electronic relaxation of pyrimidine nucleobases, nucleosides, and nucleotides in an aqueous environment generates an electronically excited intermediate state with a lifetime of tens to hundreds of picoseconds with a relatively high quantum yield (QY) of 0.2-0.5. The generation of such a long-lived excited state seems to be inconsistent with the photostability of these molecules. A recent extreme ultraviolet time-resolved photoelectron spectroscopy study by Miura et al. [J. Am. Chem. Soc. 2023, 145 (6), 3369] reinvestigated this problem and revealed that the accurately determined QY for long-lived excited states is much too low to allow an electronically excited reaction intermediate to exist. In the present study, we investigated the nature of the reaction intermediate using ultraviolet and infrared transient absorption spectroscopy, along with quantum chemical calculations to show that the intermediate is in the S0 state, and its infrared spectrum is compatible with a metastable twisted C=C species theoretically predicted by Park et al. [J. Phys. Chem. Lett. 2022, 13 (30), 7072]
Femtosecond time-resolved X-ray absorption spectroscopy of anatase TiO2 nanoparticles using XFEL
Yuki Obara, Hironori Ito, Terumasa Ito, Naoya Kurahashi, Stephan Thurmer, Hiroki Tanaka, Tetsuo Katayama, Tadashi Togashi, Shigeki Owada, Yo-ichi Yamamoto, Shutaro Karashima, Junichi Nishitani, Makina Yabashi, Toshinori Suzuki, and Kazuhiko Misawa
Struct. Dyn., 4, 044033 (2017)
The charge-carrier dynamics of anatase TiO2 nanoparticles in an aqueous solution were studied by femtosecond time-resolved X-ray absorption spectroscopy using an X-ray free electron laser in combination with a synchronized ultraviolet femtosec- ond laser (268 nm). Using an arrival time monitor for the X-ray pulses, we obtained a temporal resolution of 170 fs. The transient X-ray absorption spectra revealed an ultrafast Ti K-edge shift and a subsequent growth of a pre-edge structure. The edge shift occurred in ca. 100 fs and is ascribed to reduction of Ti by localization of gen- erated conduction band electrons into shallow traps of self-trapped polarons or deep traps at penta-coordinate Ti sites. Growth of the pre-edge feature and reduction of the above-edge peak intensity occur with similar time constants of 300–400 fs, which we assign to the structural distortion dynamics near the surface.
Ultraviolet Photochemical Reaction of [Fe(III)(C2O4)3]3- in Aqueous Solutions Studied by Femtosecond Time-Resolved X-ray Absorption Spectroscopy using an X-ray Free Electron Laser
Y. Ogi, Y. Obara, T. Katayama, Y.-I. Suzuki, S. Y. Liu, N. C-M. Bartlett, N. Kurahashi, S. Karashima, T. Togashi, Y. Inubushi, K. Ogawa, S. Owada, M. Rubesova, M. Yabashi, K. Misawa, P. Slavicek, and T. Suzuki
Struct. Dyn., 2, 034901 (2015)
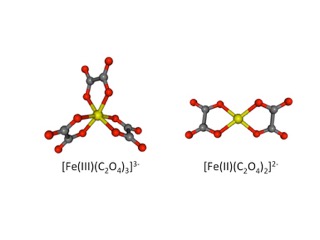
Time-resolved X-ray absorption spectroscopy was performed for aqueous ammonium iron(III) oxalate trihydrate solutions using an X-ray free electron laser and a synchronized ultraviolet laser. The spectral and time resolutions of the experiment were 1.3 eV and 200 fs, respectively. A femtosecond 268 nm pulse was employed to excite [Fe(III)(C2O4)3]3- in solution from the high-spin ground electronic state to ligand-to-metal charge transfer (LMCT) state(s), and the subsequent dynamics were studied by observing the time-evolution of the X-ray absorption spectrum near the Fe K-edge. Upon 268 nm photoexcitation, the Fe K-edge underwent a red-shift by more than 4 eV within 140 fs; however, the magnitude of the redshift subsequently diminished within 3 ps. The Fe K-edge of the photoproduct remained lower in energy than that of [Fe(III)(C2O4)3]3-. The observed red-shift of the Fe K-edge and the spectral feature of the product indicate that Fe(III) is upon excitation immediately photoreduced to Fe(II), followed by ligand dissociation from Fe(II). Based on a comparison of the X-ray absorption spectra with DFT calculations, we propose that the dissociation proceeds in two steps, forming first [(CO2•)Fe(II)(C2O4)2]2- and subsequently [Fe(II)(C2O4)2]2-.
Femtosecond time-resolved X-ray absorption spectroscopy of liquid using a hard X-ray free electron laser in a dual-beam dispersive detection method
Yuki Obara, Tetsuo Katayama, Yoshihiro Ogi, Takayuki Suzuki, Naoya Kurahashi, Shutaro Karashima, Yuhei Chiba, Yusuke Isokawa, Tadashi Togashi, Yuichi Inubushi, Makina Yabashi, Toshinori Suzuki, and Kazuhiko Misawa
Opt. Exp., 22, 1105-1113 (2014)
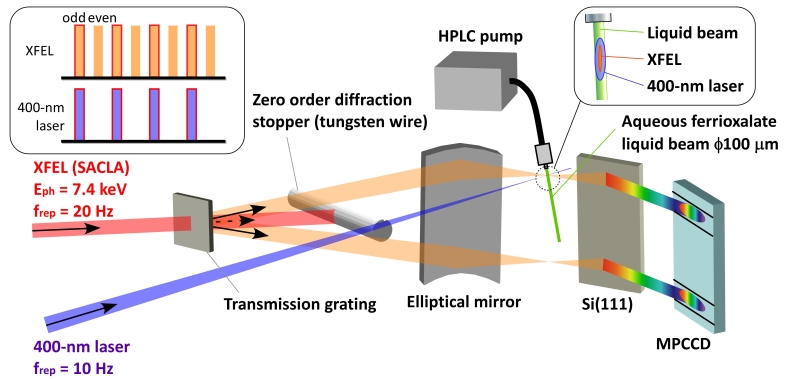
We present femtosecond time-resolved X-ray absorption spectroscopy of aqueous solution using a hard x-ray free electron laser (SACLA) and a synchronized Ti:sapphire laser. The instrumental response time is 200 fs, and the repetition rate of measurement is 10 Hz. A cylindrical liquid beam 100 mm in diameter of aqueous ammonium iron(III) oxalate solution is photoexcited at 400 nm, and the transient X-ray absorption spectra are measured in the K-edge region of iron, 7.10-7.26 keV, using a dual X-ray beam dispersive detection method. Each of the dual beams has the pulse energy of 1.4 mJ, and pump-induced absorbance change on the order of 10-3 is successfully detected. The photoexcited iron complex exhibits a red shifted iron K-edge with the appearance time constant of 260 fs. The X-ray absorption difference spectra, with and without the pump pulses, are independent of time delay after 1.5 ps up to 100 ps, indicating that the photoexcited species is long-lived.
4. Development of Ultrafast Lasers
Generation of sub-10-fs deep and extreme ultraviolet pulses for time-resolved photoemission spectroscopy
Shutaro Karashima, Chih-Jen Chen, and Toshinori Suzuki
Optics Letters, 49, 3777-3780 (2024)
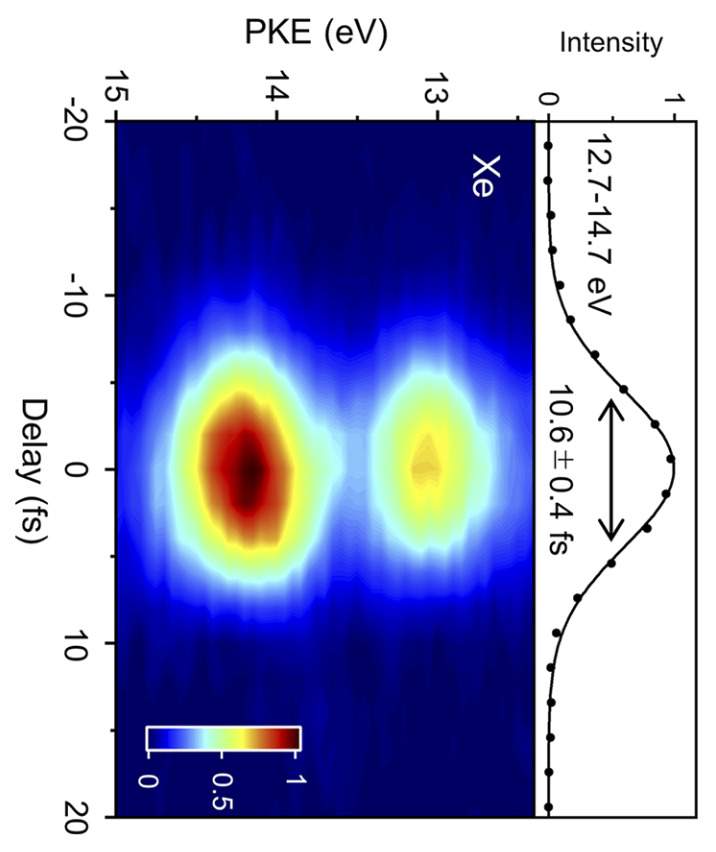
We present a light source capable of generating sub-10-fs deep UV (DUV) and extreme UV (EUV) pulses for use in time-resolved photoemission spectroscopy. The fundamental output of a Ti:sapphire laser was compressed using the multi-plate method and mixed with the uncompressed second harmonic in a filamentation four-wave mixing process to generate sub-10-fs DUV pulses. Sub-10-fs EUV pulses were generated via high-order harmonic generation driven by the second harmonic pulses that were compressed using Ar gas and chirped mirrors. The minimum cross correlation time between 267 and 57 nm (corresponding to 21.7 eV) was measured to be 10.6 ± 0.4 fs.
UV-driven harmonic generation for time-resolved photoelectron spectroscopy of polyatomic molecules
Shunsuke Adachi, and Toshinori Suzuki
Appl. Sci., 8, 1784 (2018)
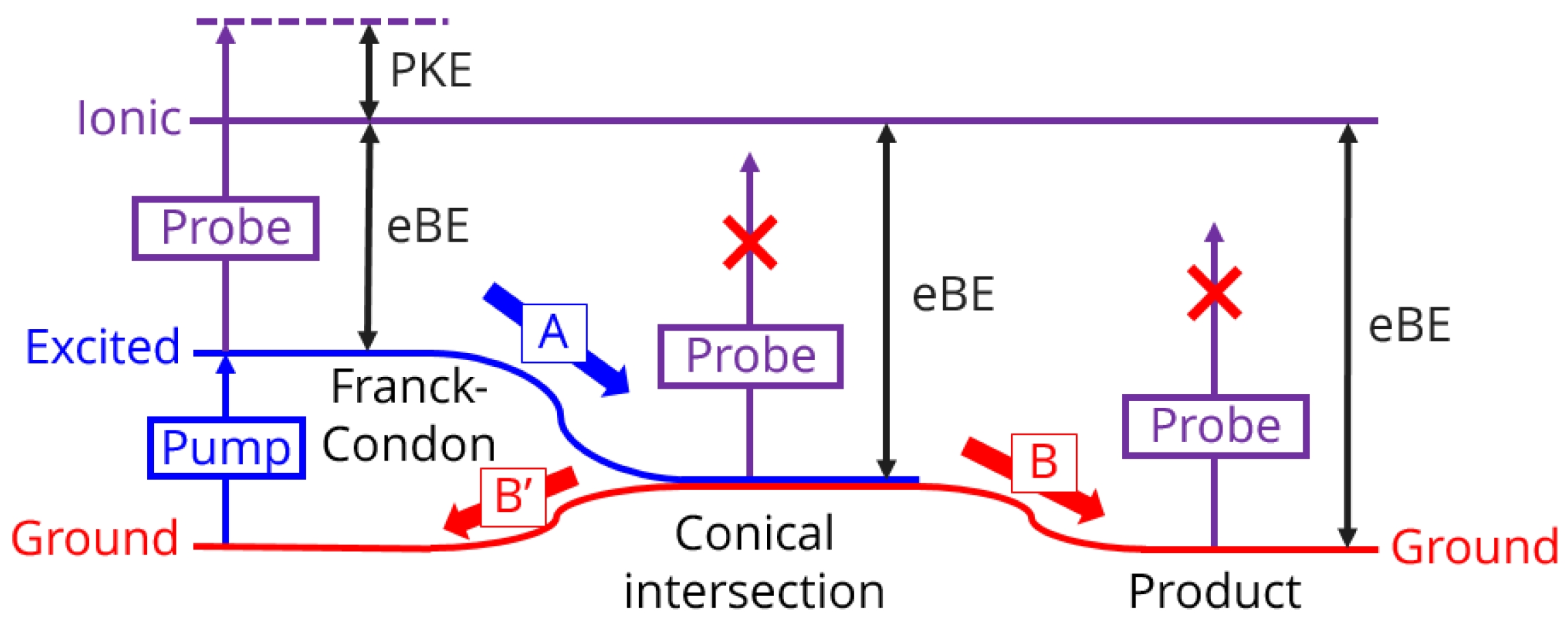
A single-order harmonic pulse in the vacuum-ultraviolet (VUV) is highly desirable for time-resolved photoelectron spectroscopy (TRPES) of polyatomic molecules. A high-power 9th harmonic of a Ti:sapphire laser (hv = 14 eV) is obtained using a UV driving laser at 270 nm (the 3rd harmonic). We describe our recent efforts to develop VUV TRPES combined with UV-driven harmonic generation, and present a few representative results from our recent TRPES studies.
Probing ultrafast spin-relaxation and precession dynamics in a cuprate Mott insulator with seven-femtosecond optical pulses
T. Miyamoto, Y. Matsui, T. Terashige, T. Morimoto, N. Sono, H. Yada, S. Ishihara, Y. Watanabe, S. Adachi, T. Ito, K. Oka, A. Sawa, and H. Okamoto
Nat. Commun., 9, 3948 (2018)

A charge excitation in a two-dimensional Mott insulator is strongly coupled with the surrounding spins, which is observed as magnetic-polaron formations of doped carriers and a magnon sideband in the Mott-gap transition spectrum. However, the dynamics related to the spin sector are difficult to measure. Here, we show that pump-probe reflection spectroscopy with seven-femtosecond laser pulses can detect the optically induced spin dynamics in Nd2CuO4, a typical cuprate Mott insulator. The bleaching signal at the Mott-gap transition is enhanced at ~18 fs. This time constant is attributable to the spin-relaxation time during magnetic-polaron formation, which is characterized by the exchange interaction. More importantly, ultrafast coherent oscillations appear in the time evolution of the reflectivity changes, and their frequencies (1400–2700 cm−1) are equal to the probe energy measured from the Mott-gap transition peak. These oscillations can be interpreted as the interference between charge excitations with two magnons originating from charge–spin coupling.
Time-resolved photoelectron spectroscopy of polyatomic molecules using 42-nm vacuum ultraviolet laser based on high harmonics generation
Junichi Nishitani, Christopher W. West, Chika Higashimura, and Toshinori Suzuki
Chem. Phys. Lett., 684, 397-401 (2017)
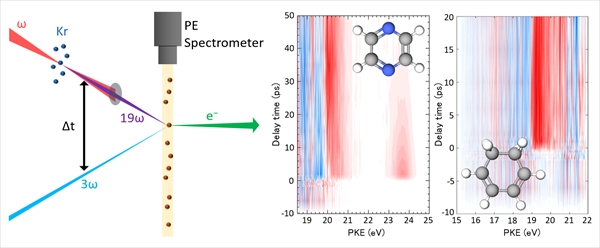
Time-resolved photoelectron spectroscopy (TRPES) of gaseous polyatomic molecules using 266-nm (4.7 eV) pump and 42-nm (29.5 eV) probe pulses is presented. A 1-kHz Ti:sapphire laser with a 35 fs pulse duration is employed to generate high harmonics in Kr gas, and the 19th harmonic (42-nm) was selected using two SiC/Mg mirrors. Clear observation of the ultrafast electronic dephasing in pyrazine and photoisomerization of 1,3-cyclohexadiene demonstrates the feasibility of TRPES with the UV pump and VUV probe pulses under weak excitation conditions in the perturbation regime.
Few-cycle pulse generation from noncollinear optical parametric amplifier with static dispersion compensation
Shunsuke Adachi, Yuya Watanabe, Yuki Sudo, and Toshinori Suzuki
Chem. Phys. Lett., 683, 7-11 (2017)
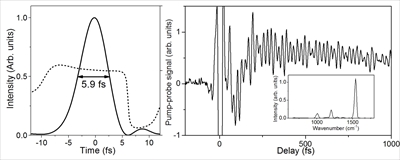
We present a novel design of a few-cycle noncollinear optical parametric amplifier (NOPA) pumped by the second harmonic of a Ti:sapphire laser. A quasi-transform-limited sub-6 fs pulse width was realized by static dispersion compensation with commercially available chirped mirrors. The performance of the NOPA was tested by performing transient absorption spectroscopy on sensory rhodopsin II, and we observe short-lived oscillatory components that are associated with the vibrational coherence from the isomerizing molecule in the excited electronic state.
Self-compression of femtosecond deep-ultraviolet pulses by filamentation in krypton
Shunsuke Adachi, and Toshinori Suzuki
Opt. Lett., 42, 1883-1886 (2017)
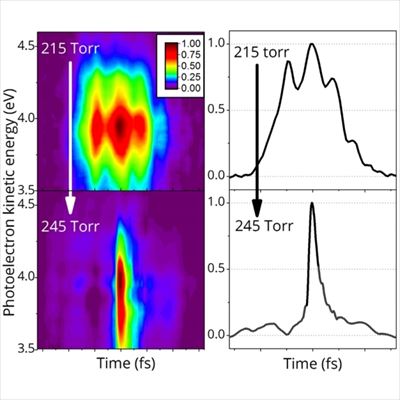
We demonstrate self-compression of deep-ultraviolet (DUV) pulses by filamentation in krypton. In contrast to self-compression in the near-infrared, that in the DUV is associated with a red-shifted sub-pulse appearing in the pulse temporal profile. The achieved pulse width of 15 fs is the shortest among demonstrated sub-mJ deep-ultraviolet pulses.
Pump-probe photoelectron spectroscopy by a high-power 90 nm vacuum-ultraviolet laser
Motoki Sato, Yoshi-ichi Suzuki, Toshinori Suzuki, and Shunsuke Adachi
Appl. Phys. Express, 9, 022401 (2016)
We present pump.probe photoelectron spectroscopy of Kr and NO using a high-power vacuum-ultraviolet (VUV) laser at a wavelength of 90 nm. Clear quantum beats are observed in the photoelectron angular distributions as well as in the photoelectron yields, resulting from the coherent excitation of two Kr Rydberg states by the VUV pump. The entire Franck.Condon envelope of the NO A(2Σ+) excited state is also successfully captured by the VUV probe.
Generation of sub-17 fs vacuum ultraviolet pulses at 133 nm using cascaded four-wave mixing through filamentation in Ne
Takuya Horio, Roman Spesyvtsev, and Toshinori Suzuki
Opt. Lett., 39, 6021-6024 (2014)
The sixth harmonic (6ω, 133 nm) of a Ti:sapphire laser is generated using cascaded four-wave mixing in filamentation propagation of the fundamental (ω) and the second harmonic (2ω) pulses through Ne gas. The method provides the 6ω pulse energy higher than 5 nJ/pulse at 1 kHz and a pulse duration shorter than 17 fs without dispersion compensation.
Simultaneous generation of sub-20 fs deep and vacuum ultraviolet pulses in a single filamentation cell and application to time-resolved photoelectron imaging
Takuya Horio, Roman Spesyvtsev, and Toshinori Suzuki
Opt. Exp., 21, 22423-22428 (2013)
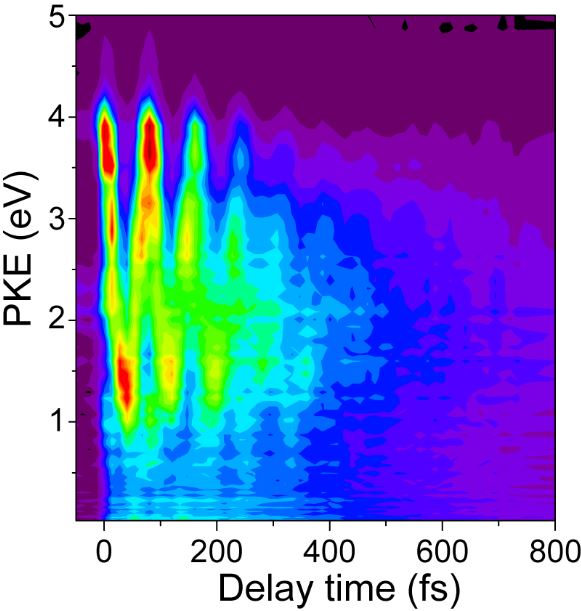
Sub-20 fs pulses of the third, fourth, and fifth harmonics of a Ti:sapphire laser are simultaneously generated using cascaded four-wave mixing in filamentation propagation of the fundamental frequency and the second harmonic pulses in Ne gas. Reflective optics under vacuum are employed after the four-wave mixing to minimize material dispersion of the optical pulses. The cross-correlation between 198 and 159 nm pulses of 18 fs is achieved without dispersion compensation. This new light source is applied to time-resolved photoelectron imaging of carbon disulfide (CS2).
Generation of intense single-order harmonic pulse in the vacuum ultraviolet region using a deep ultraviolet driving laser
Shunsuke Adachi, Takuya Horio, and Toshinori Suzuki
Opt. Lett., 37 (11), pp. 2118-2120 (2012)

A 90 nm single-order harmonic pulse with a 200 nJ on-target pulse energy at 1 kHz was realized through a harmonic generation process with a 35 fs Ti:Sa third harmonic in a Kr gas cell.
Spectral phase transfer to ultrashort UV pulses through four-wave mixing
Peng Zuo, Takao Fuji, and Toshinori Suzuki
Opt. Exp., 18 (15), 16183-16192 (2010)

Transfer of spectral phase from near infrared ultrashort pulses to deep ultraviolet (UV) sub-30-fs pulses through four-wave mixing process is demonstrated. Micro joule UV pulses at 237 nm were generated by nonlinear mixing of second harmonic pulses of Ti:sapphire laser output and near infrared pulses from a noncollinear optical parametric amplifier. Chirp of the near infrared pulse was transfered to the UV pulse with the opposite sign. A positively chirped near infrared pulse was used for generating a negatively chirped UV pulse, which was compressed down to 25 fs by a magnesium fluoride window.