"Developing new theoretical frameworks and uncovering previously uncharted phenomena"-this is the guiding philosophy of our research.
The dimensionality of the electronic wavefunction grows exponentially with the number of electrons. For many stable organic molecules, however, the wavefunction possesses a relatively simple internal “structure,” allowing sophisticated single-reference theories to solve the Schrödinger equation within a compact subspace. Yet, once we step beyond this familiar regime, we encounter the domain of multireference problems –regions where standard theories become unreliable or even fail outright.
Our group develops new wavefunction theories to navigate these unexplored areas of chemical electronic structure. We apply these methods to phenomena in which strong multireference character is essential, including bond dissociation, excited states, and transition-metal complexes.
Development of Wavefunction Theories for Strongly Correlated Electrons
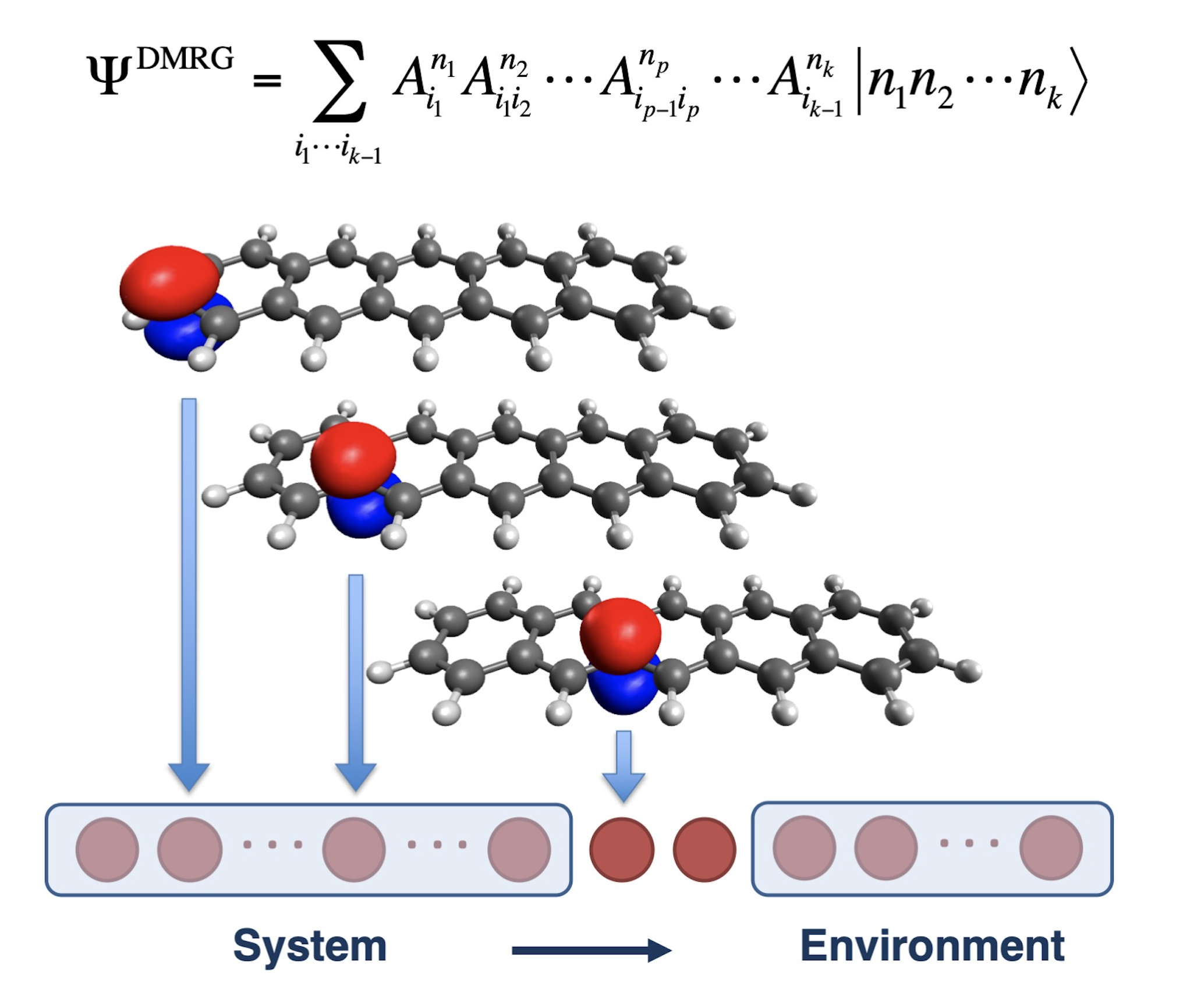 Systems exhibiting strong electron correlation—so-called multireference electronic states—remain among the most difficult challenges in modern quantum chemistry.
In these systems, the wavefunction does not possess an easily identifiable structure, making dimensional reduction extremely hard.
Consequently, computational cost grows exponentially with valence-electron count, leaving many important chemical problems unsolved even today.
Systems exhibiting strong electron correlation—so-called multireference electronic states—remain among the most difficult challenges in modern quantum chemistry.
In these systems, the wavefunction does not possess an easily identifiable structure, making dimensional reduction extremely hard.
Consequently, computational cost grows exponentially with valence-electron count, leaving many important chemical problems unsolved even today.
To overcome these limitations, we develop new multireference formalisms based on tensor-network wavefunctions. Using these methods, we investigate transition-metal complexes, π-conjugated molecules (particularly under photoexcitation), and molecular spin systems—domains where strong correlation plays a central role.
Transition-Metal Complexes
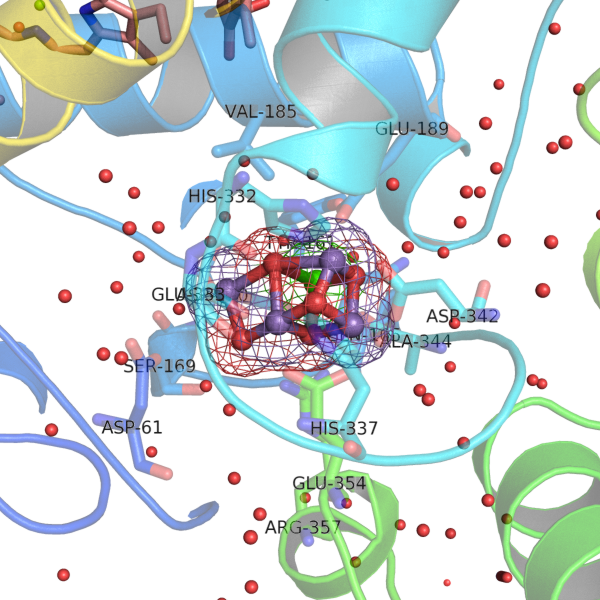 The chemical bonding between transition-metal ions and ligands is often portrayed as simple “coordination bonds,”
yet accurate wavefunction calculations reveal remarkable complexity.
Even basic notions like “oxidation state” become ambiguous, and 4d orbitals can acquire partial occupation even for 3d ions such as Fe or Mn.
The chemical bonding between transition-metal ions and ligands is often portrayed as simple “coordination bonds,”
yet accurate wavefunction calculations reveal remarkable complexity.
Even basic notions like “oxidation state” become ambiguous, and 4d orbitals can acquire partial occupation even for 3d ions such as Fe or Mn.
These observations remind us that the molecular orbital picture—central to modern chemistry—is ultimately only an approximate model. Since no definitive theoretical framework exists for these systems, theoretical chemists must navigate largely unexplored territory. We embrace this complexity, as it is the origin of fascinating functions such as catalysis and photochemical activity, and we use state-of-the-art theory to unravel their electronic structures.
Excited States
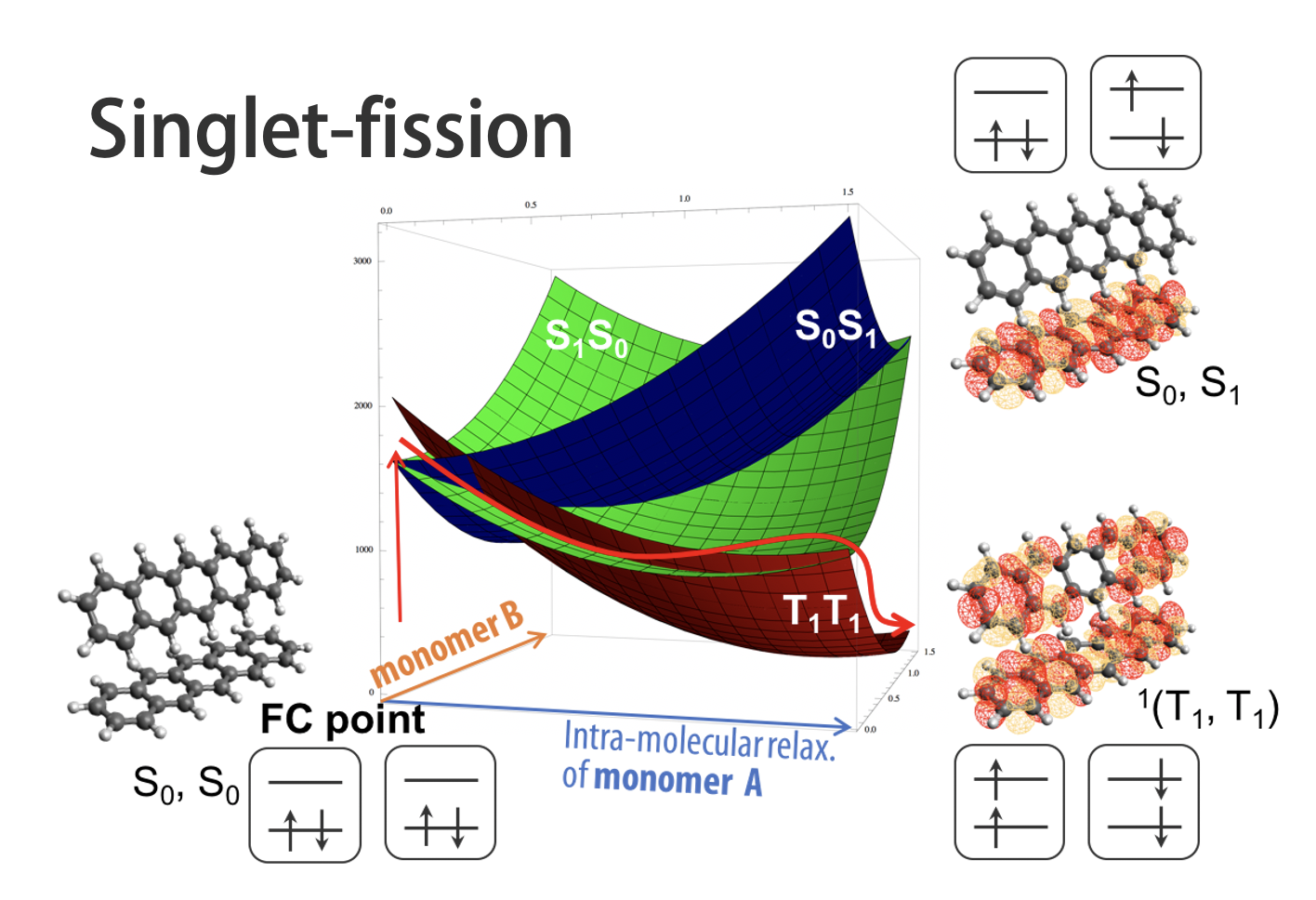 When molecules absorb sunlight or laser light, they transition into electronically excited states—states fundamentally more complex than ground states.
Predicting photochemical outcomes solely from hand-drawn structural formulas is essentially impossible.
When molecules absorb sunlight or laser light, they transition into electronically excited states—states fundamentally more complex than ground states.
Predicting photochemical outcomes solely from hand-drawn structural formulas is essentially impossible.
From a theoretical perspective, treating all possible excited states without bias requires an enormous wavefunction space, which is practically unattainable. Therefore, careful physical insight is necessary to construct an appropriate active space and interpret results, often through iterative analysis and refinement.
The very difficulty of these problems reflects the essence of chemical phenomena—emergent behavior governed by delicate energetic balances.
Molecular Spin Systems
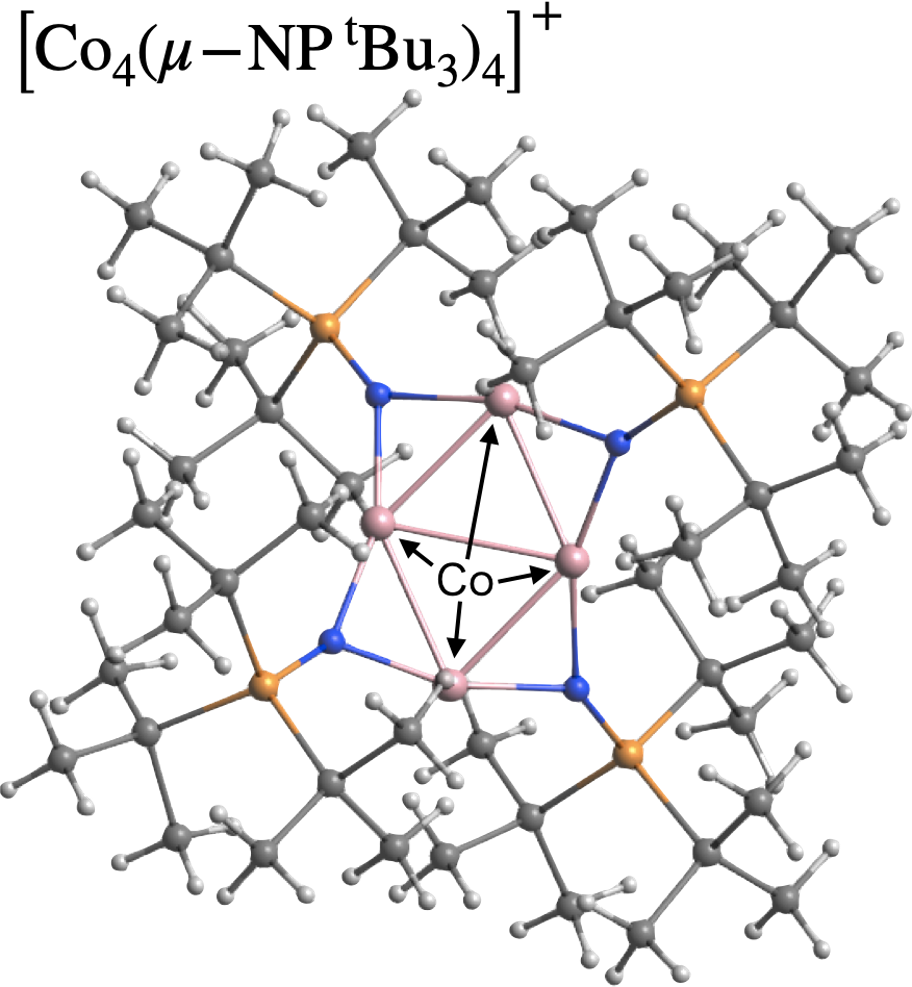 Electron spin is an indispensable degree of freedom in chemistry.
In most molecules the ground state is a singlet with no net spin polarization,
making spin easy to overlook.
However, to realize molecular functionalities such as switching, control, or quantum information processing,
spin must be treated as a crucial, tunable variable.
Electron spin is an indispensable degree of freedom in chemistry.
In most molecules the ground state is a singlet with no net spin polarization,
making spin easy to overlook.
However, to realize molecular functionalities such as switching, control, or quantum information processing,
spin must be treated as a crucial, tunable variable.
Our group focuses particularly on spin–orbit coupling arising from relativistic effects, and aims to design molecules for single-molecule magnets, spin polarization, and efficient utilization of triplet energies. A major goal is to discover “languages”—conceptual frameworks—that bridge electronic-level angular-momentum physics with chemically meaningful molecular design principles.
Algorithm Development for Quantum Computers
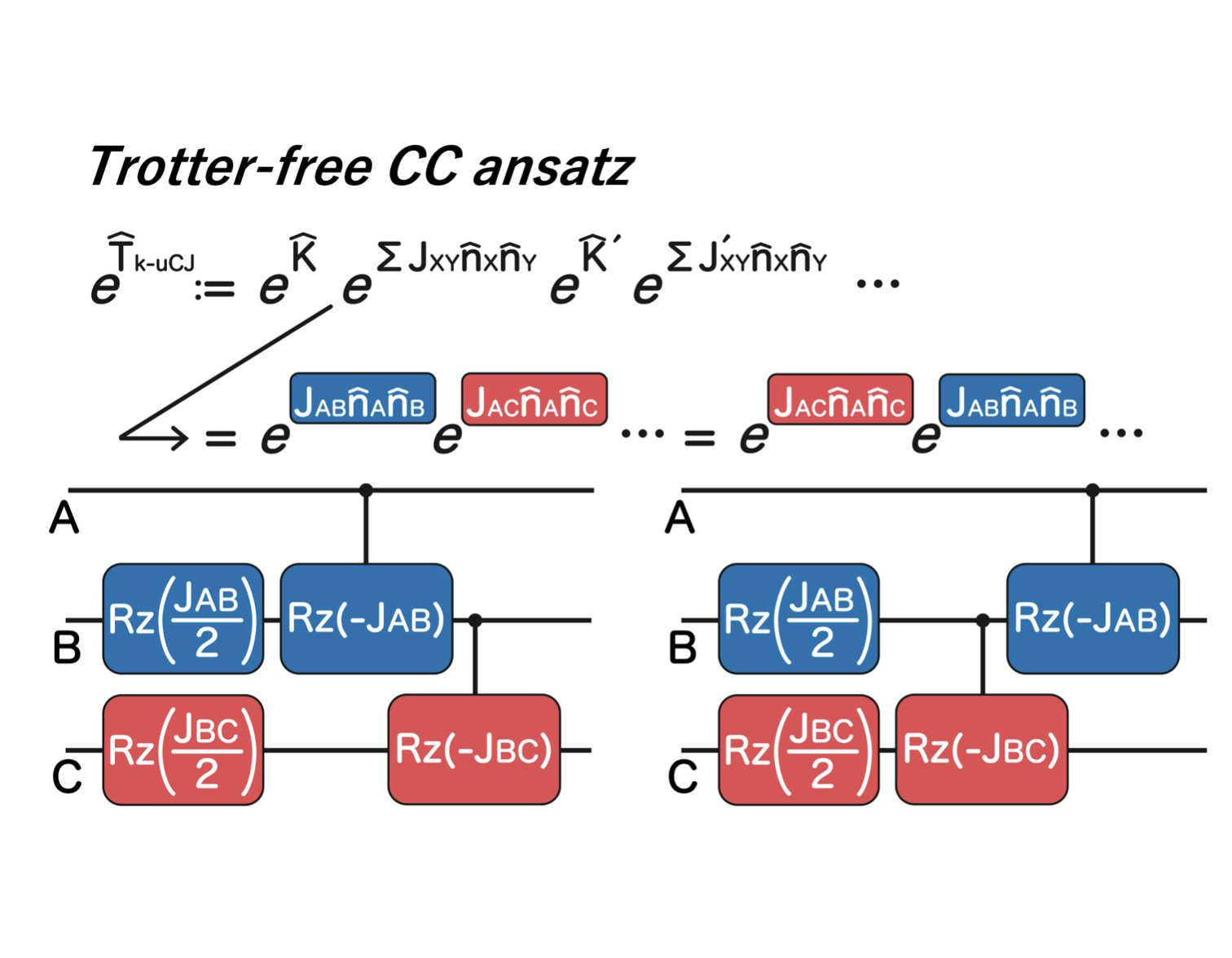 Quantum chemistry is expected to be one of the first scientific domains profoundly transformed by practical quantum computers.
Although the timeline to large-scale quantum hardware remains uncertain,
the theoretical landscape has opened new possibilities.
Quantum chemistry is expected to be one of the first scientific domains profoundly transformed by practical quantum computers.
Although the timeline to large-scale quantum hardware remains uncertain,
the theoretical landscape has opened new possibilities.
Methods once dismissed as impractical on classical computers may become viable in a quantum environment. This has revitalized foundational discussions in quantum chemistry and created opportunities for conceptual breakthroughs.
We aim to develop quantum algorithms with flexibility and creativity—never losing the sense of play— in the hope that they will open new frontiers in electronic-structure theory.